22 Laboratory aspects of blood transfusion
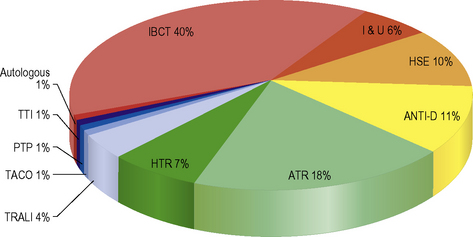
Safe and effective blood transfusion requires the combined efforts of blood transfusion services, biomedical scientists and clinicians to ensure the highest standards are applied to all the systems in a complex process from ‘vein to vein’. This chapter provides a description of the laboratory framework required to provide the right blood components to the right patients at the right time. The increased awareness of what can go wrong with blood transfusion comes from a number of sources including the Serious Hazards of Transfusion UK haemovigilance scheme, which was started in 1996.1,2 This confidential reporting scheme, using detailed root cause analysis of errors, has provided data that have informed both national bodies and local transfusion services of measures to introduce in order to reduce risk (Fig. 22.1). It is clear that multiple errors can contribute to a single adverse event and that many of these are outside the control of the transfusion laboratory.
Within the laboratory setting, the application of strict protocols for sample labelling and testing, robust laboratory procedures, reliable documentation, frequent staff training and competency assessment should be used. Recently, the UK Transfusion Collaborative, comprising representatives of the professional bodies involved in UK transfusion practice, has produced some recommendations to reduce laboratory errors in transfusion. These address issues such training, competency, staffing levels and automated systems.3
This chapter is concerned with the testing of patient samples prior to the provision of appropriate compatible blood components including identification of red cell antibodies. It also covers compatibility testing and investigation of transfusion reactions and the testing required in other special situations including the antenatal and postnatal settings. National professional bodies such as the British Committee for Standards in Haematology (BCSH)4 and the AABB5 issue guidance to transfusion laboratories and this has been referenced where appropriate.
There is a new regulatory framework governing hospital transfusion laboratory practice which was implemented after the publication of two European Union Directives: 2002/98/EC and 2004/33/EC.6 In the UK these are the Blood Safety and Quality Regulations 2005 (Statutory Instruments 2005/50, 2005/1098 and 2006/2013) and they set standards for quality and safety of human blood and blood components in hospital ‘blood banks’ as well as ‘blood establishments’ (the UK Blood Services).7 ‘Blood banks’ are regulated in their roles of storing, distributing and performing compatibility tests, on blood and blood components for use in hospitals. The implications for hospital transfusion practice are the requirement for ‘vein to vein’ traceability of blood components, the importance of maintaining the ‘cold chain’ for all therapeutic blood components, the need to store transfusion records for 30 years and the requirement for a quality management system.6,7 UK laboratories have to assess themselves and submit an annual compliance report to the competent authority, which is the Medicines and Healthcare Products Regulatory Agency (MHRA).8 The MHRA carries out laboratory inspections to assess compliance with these regulations.
The UK government, through the National Blood Transfusion Committee, continues to promote the safe and effective transfusion of blood components and has published three Health Service Circulars.9 These documents aim to ensure that a team approach to blood transfusion safety is taken via the local clinical governance arrangements and to promote good transfusion practice via Hospital Transfusion Committees and Hospital Transfusion Teams supported by Regional Transfusion Committees.
Technology and automation in blood transfusion laboratories
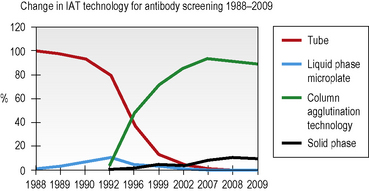
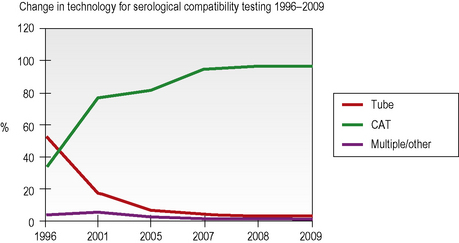
Column agglutination (CAT) and solid-phase technology can both be used on automated machines and CAT can also be used by manual techniques. In UK hospital transfusion laboratories these technologies have replaced tube techniques and liquid-phase microplates for antibody screening and crossmatching (Figs 22.2, 22.3).10
Individual laboratories need to make careful and informed decisions when selecting reagents for pre-transfusion testing. It is vital that any abbreviated testing in an automated or semiautomated system is carefully evaluated for the risks that could ensue if important controls were omitted when using this technology for blood grouping.11
Laboratory information management systems (LIMS) store patient details and results of laboratory tests, allowing timely and accurate access to important information. In the transfusion department, IT systems have a much broader use and the updated BCSH guidelines for the specification and use of Information Technology (IT) systems in blood transfusion practice (2006) reflect this.12 Where possible, using bidirectional or unidirectional interfaces to automated blood grouping analysers, IT systems are used to eliminate errors that can arise when a manual step is employed, including interpretation of test results.
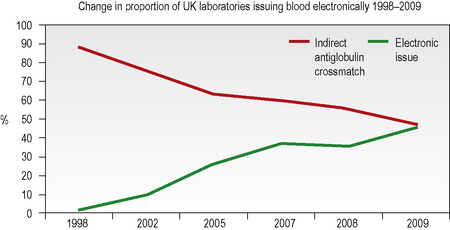
Computer algorithms support the ‘electronic issue’ of blood to patients with a negative antibody screen without the need to perform an antiglobulin crossmatch and, in the UK in 2009, 46% of laboratories were using this system for some or all of their patients (Fig. 22.4). The use of automation for all aspects of compatibility testing is now recommended practice as it is recognized that it is safer.3 Automation brings several or all of the discrete activities of compatibility testing into a single platform process. It provides various levels of increased security over manual testing and may provide justification for abbreviated pre-transfusion testing (e.g. abandoning duplicate D, previously termed ‘RhD’, typing or reverse ABO grouping in the presence of a valid historical group). A risk assessment must be made and documented prior to any abbreviation of an established procedure, with consideration being given to the presence or absence of key functions in the automated equipment. The BCSH guidelines for compatibility procedures12 and guidance from the MHRA13 give a list of factors to be taken into consideration and the reader is advised to consult these prior to implementing automated or semiautomated systems.
Pre-transfusion compatibility systems
Documentation of the Transfusion Process
All stages of the transfusion process must be clearly documented and these records must be kept. In addition to guidance from the UK Royal College of Pathologists on the retention of documents,18 the BSQR 2005 regulations stipulate that the records must be accessible for 30 years.7 This allows any blood component to be traced from the donor to the recipient should information come to light about any potential infective risks to the recipient.
Computer records are easier to search than paper records, but laboratory information systems are likely to become obsolete and be replaced several times within this mandatory 30-year period, so provision must be made to store historical data in an accessible format when procuring a replacement computer system.12 Patient-held records are useful for patients who are treated in more than one institution, particularly if they have red cell antibodies and require phenotyped blood or if they have special requirements because of their underlying disease or its treatment. Credit card-sized records with corresponding patient information leaflets are issued by some transfusion centres to patients with red cell antibodies and similar cards exist for patients who require irradiated cellular blood components.19
Identification and Storage of Blood Samples
On being received in the laboratory, the details on the request form must be checked against the blood sample. Each blood sample must be labelled with a unique sample number. Barcode labels offer the advantage of positive sample identification and reduce the number of transcription errors. Samples inadequately or inaccurately labelled should NOT be used for pre-transfusion testing.11
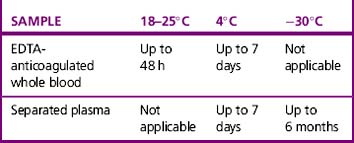
Whole blood samples should be tested as soon as possible because they will deteriorate over time. Problems associated with storage include lysis of the red cells, loss of complement in the serum and decrease in potency of antibodies. The BCSH guidelines11 indicate working limits as outlined in Table 22.1. If separated plasma or serum samples are stored for later serological crossmatch, care must be taken to ensure that the patient has not been transfused in the interim. It has been recommended that samples should be kept for a minimum of 7 days from group and screen, stored at 4°C. Samples should be retained post-transfusion for investigation of acute transfusion reactions and preferably stored for 7 days post-transfusion to enable investigation of delayed transfusion reaction.18
ABO and D grouping
ABO Grouping
This is an excellent built-in check for the ‘forward’ or cell group and has always been considered to be an integral part of ABO grouping, allowing the reading and recording of test results to be split into two discrete tasks. However, with secure, fully automated systems, linked to secure laboratory information management systems that, in combination, have the ability to prevent procedural ABO grouping errors, some laboratories now omit the reverse group when testing samples for which a historical group is available.11 This should only be considered following a careful risk assessment and taking into account that the first sample taken may have been from the wrong patient. This may be in the order of 1:2000 samples.16 Any discrepancy between the forward and reverse groups should be investigated further and any repeat tests should be undertaken using cells taken from the original sample rather than from a prepared cell suspension.
D Grouping
Reagents for D Grouping
DVI is the partial D with the fewest epitopes; therefore of all the D variants, DVI individuals are those most likely to form anti-D and a case of severe haemolytic disease of the fetus and newborn (HDFN) has been described.20 For this reason, anti-D monoclonal reagents that do not detect DVI should be selected for testing patients’ samples.21,22 The use of anti-CDE reagents has led to the misinterpretation of r′ and r″ cells in UK National External Quality Assessment Scheme (NEQAS) exercises and, because they are of no value in routine patient typing, their use is not recommended.10,11 Selection of high-avidity monoclonal anti-D reagents will allow detection of all but the weakest examples of weak D, negating the need to use more sensitive techniques to check the D status of apparent D negatives.
Methods
There are several techniques available for routine ABO and D grouping including tube test, slide test, liquid-phase and solid-phase microplates and columns. Other techniques for blood grouping have been described but they are not in routine use. For example, molecular ABO typing is reserved for investigating anomalous ABO groups, in organ transplantation where red cells from the donor are not available, forensic practice and paternity testing.23 Care should be taken to use the appropriate reagent because not all reagents have been validated by the manufacturer for all techniques.
EDTA for diluents
Liquid-phase microplate methods
Liquid-phase microplate technology provides a cheap and secure method for batch testing when semiautomation is utilized for dispensing and reading but it is no longer the grouping technique of choice in the UK (see Fig. 22.2). In 2009, a UK NEQAS survey showed that only 13% of responding laboratories were using microplates for grouping, down from 41% in a similar survey in 2002.10
The plate is usually laid out as 12 × 8 (12 tests and 8 reagents) but may be used in the opposite orientation (8 × 12), depending on the number of reagents and controls required. Particularly if performing this technique manually, the anti-D reagents should be kept away from the anti-A and anti-B reagents because splashing between wells can occur when dispensing reagents and handling the plates during testing if insufficient care is taken. The following method is recommended:24
Column agglutination techniques
CAT techniques (see Chapter 21, p. 502) are now the commonest method for grouping in the UK (80% of laboratories who responded to a UK NEQAS survey in 2009, see Fig. 22.2), especially where automated systems are in place; these should always be performed in accordance with the manufacturer’s instructions. There are several different profiles to choose from and some cards/cassettes include monoclonal antibodies to other blood group antigens (e.g. K) in addition (see Chapter 21, p. 502 and Fig. 22.6). Forward and reverse grouping may be undertaken in separate cards/cassettes.
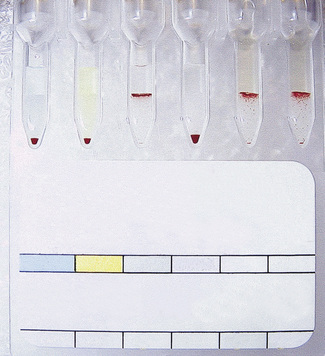
Controls
Positive and negative controls should be included with every test or batch of manual tests. In fully automated systems, the controls should be set up at least twice in a 24-h period. The timings should coincide with machine startup and changing of reagents, taking account of the length of time that reagents have been kept at room temperature on the machine. The control samples should be loaded in the same way as the test samples. The required controls are shown in Table 22.2. Where controls do not give the expected reactions, investigations should be undertaken to determine the validity of all tests undertaken subsequent to the most recent valid control results.
Table 22.2 Control cells for blood grouping
| Reagent | Positive control | Negative control |
|---|---|---|
| Anti-A | A cells | B cells |
| Anti-B | B cells | A cells |
| Anti-D | D positive cells | D negative cells |
| A1 cells | Anti-A | Anti-B |
| B cells | Anti-B | Anti-A |
Causes of Discrepancies in ABO/D Grouping
T-activation/polyagglutination
Polyagglutination25 describes agglutination of red cells by all or most normal adult sera but not by the patient’s own serum. This is as the result of IgM antibodies reacting with an antigen on the red cells which is usually hidden but can be exposed by enzyme activity. The most common form is T-activation, which occurs when the bacterial enzyme neuraminidase cleaves N-acetyl neuraminic acid from the red cell membrane, exposing the T antigen.
Acquired B
The acquired B antigen is usually caused by a bacterial deacetylase enzyme acting on A1 red cells and producing a B-like substance. Some anti-B reagents react strongly with the acquired B antigen (e.g. those derived from the ES4 clone).26 Such anti-B reagents are rare but should be avoided in routine blood grouping.
Potentiators
Red cells may be coated with IgG as a result of in vivo sensitization. The use of potentiated techniques, such as the antiglobulin test for D typing, or of potentiated reagents for ABO or D typing, may result in a false-positive reaction; the latter would also result in a positive reaction with the diluent control, but UK NEQAS data have shown that some laboratories fail to include an appropriate control or fail to understand the significance of a positive control.27 For this reason, use of potentiated techniques or reagents for blood grouping is not advised.
False-Negative Reactions
D variant phenotypes
Weak D phenotypes are where the entire D antigen is present but there are fewer D antigen sites per cell and most weak D types group as D positive with the currently available high-avidity commercial monoclonal anti-D reagents. Where differing reactions are obtained with two reagents, the patient may be a partial D (i.e. one or more of the epitopes of the D antigen is missing). It was generally thought that patients who are weak D are unable to make anti-D and may be treated as D positive whereas some patients with partial D may be capable of making immune anti-D following sensitization with the missing epitope. This concept has been challenged by reports of weak D patients with anti-D.28
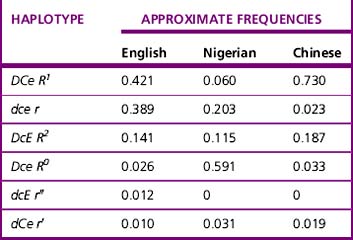
Current advice about choice of D-typing reagents is that because DVI lacks the most epitopes, such individuals are likely to make anti-D when challenged by transfusion or pregnancy. For this reason, anti-D reagents for routine grouping of patients’ samples should not detect DVI.11 There is little evidence to suggest that a DVI donor would elicit an immune response in a recipient who is D negative; however, weak D positive and partial D donors, including DVI donors, should be classified as D positive.29 There are some important ethnic differences in the frequency of different Rh haplotypes, as shown in Table 22.3. Resolution of anomalous D grouping where a partial D is suspected now includes both serological testing and genotypic studies.30
Antibody screening
Red Cell Reagents
In the UK, the following antigens should be expressed as a minimum: C, c, D, E, e, K, k, Fya, Fyb, Jka, Jkb, S, s, M, N and Lea; one cell should be R2R2 and another R1R1 or R1wR1. The following phenotypes should also be represented in the screening set: Jk(a+b−), Jk(a−b+), S+s−, S−s+, Fy(a+b−) and Fy(a−b+) (Table 22.4). These recommendations for homozygosity are based on UK data regarding the incidence of delayed haemolytic transfusion reactions, the need for high sensitivity in the detection of Kidd antibodies and the poorer performance of column agglutination techniques in the detection of some examples of Kidd antibodies using heterozygous cells.11,29 The requirement for the expression of Cw and Kpa antigens on screening cells has been the cause of much debate but in the UK and the USA detection of anti-Cw or anti-Kpa is not a requirement even in the absence of an antiglobulin crossmatch.11,29 This is because these are low-frequency antigens and the antibodies rarely cause delayed haemolytic transfusion reactions or severe haemolytic disease of the newborn.
Table 22.4 Expression of red cell antigens on screening cells11
| Blood group system | Antigen | Homozygous cells recommended |
|---|---|---|
| Rh | C | Yes (R1R1 or R1wR1) |