Ionizing radiation
David J. Grdina, PhD
Overview
Many experimental and epidemiologic studies have since confirmed the oncogenic effects of radiation. This chapter reviews briefly the effects of ionizing radiation on biological systems, adaptive and bystander effects, cellular and molecular mechanisms for radiation carcinogenesis, and pharmacologic countermeasures that can mitigate against these processes.
Development of radiation injury
The hazards of exposure to ionizing radiation were recognized shortly after Roentgen’s discovery of the X-ray in 1895. Acute skin reactions were observed in many individuals working with early X-ray generators, and by 1902, the first radiation-induced cancer was reported arising in an ulcerated area of the skin. Within a few years, a large number of such skin cancers had been observed, and the first report of leukemia in five radiation workers appeared in 1911.1 Figure 1 describes the interaction of ionizing radiation with biologic tissues and the development of radiation injury. The ionizing event involves the ejection of an orbital electron from a molecule, producing a positively charged or “ionized” molecule. These molecules are highly unstable and rapidly undergo chemical change. This change results in the production of free radicals. The most common products of this process are the result of the decomposition of water giving rise to both superoxide anion (O2−) and hydrogen peroxide (H2O2) and the highly reactive hydroxyl radical, which has as a result a very short life span and can diffuse only on the average about 4 nm before reacting with other molecules.2 Reactive oxygen species (ROS) production can be amplified by intermitochondrial communication that results in a subsequent magnification of the ROS damage signal.3 The process of “ROS-induced ROS release,” or RIRR is one potential mechanism for this. Under conditions of an excessive oxygen stress burden, the increase in ROS within the mitochondria reaches a threshold that triggers the opening of either the mitochondrial permeability transition (MPT) pore or the inner membrane anion channel (IMAC), which in turn leads to the simultaneous collapse of mitochondrial membrane potential and a transient increase in ROS generation by the electron transfer chain. Release of this ROS burst into the cytosol functions as a second messenger to activate RIRR in neighboring mitochondria.4, 5 Radiation can produce a transient generation of reactive oxygen or nitrogen within minutes accompanied by a reversible depolarization of mitochondrial membrane potential.3 Radiation damage in a single mitochondrium can be transmitted via a reversible Ca2+-dependent MPT to adjacent mitochondria resulting in the amplification of ROS generation. ROS amplification and propagation resulting from such a cascade can then damage important biological targets such as DNA, the nuclear matrix, cytoplasmic transport mechanisms, and both mitochondrial and cellular membranes resulting in cell death. The ROS amplification process may also be interfered by endogenous antioxidants such as superoxide dismutases (SODs) or exogenously added antioxidants.
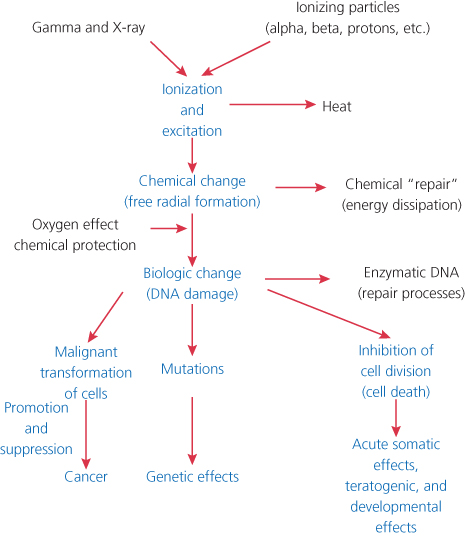
Figure 1 Development of radiation injury.
Principal cellular and tissue effects of radiation
Cell killing
Radiation can kill cells by apoptosis and interphase death.6–8 Cells undergoing apoptosis usually die in interphase within a few hours of irradiation. Apoptotic cell death can be induced by exposure to relatively low doses of radiation6 and be a significant cause of death in hematopoietic or lymphoid cells exposed to higher radiation doses.
Radiation-induced apoptosis is dependent upon both the functional activity of the p53 gene as well as p53-independent pathways9 all of which converge on the activation of proteases called caspases.10 It has been proposed that p53-dependent apoptosis may involve the transcriptional induction of redox-related genes with the formation of ROS, leading to cell death by oxidative stress.11
The second mechanism for cell killing is radiation-induced reproductive failure. The inhibition of cellular proliferation is the mechanism by which radiation kills most mammalian cells. Symptoms of acute exposure to whole-body irradiation in human beings are usually observed only following doses of 100 cGy or greater.
Mutagenesis
Studies of the induction of single-gene mutations in human cells have been limited to several genetic loci. Of particular note is the X-chromosomal gene for hypoxanthine-guanine phosphoribosyltransferase (HPRT).12, 13 The induction of mutations in human cells is a linear function of dose down to doses as low as 10 cGy, and perhaps as low as 1 cGy, and the dose-rate effect appears to be relatively small.14, 15 DNA structural analyses show that the majority of radiation-induced mutations in human cells result from large-scale genetic events involving loss of the entire active gene and often extending to other loci on the same chromosome.16
The major potential consequence of radiation-induced mutations in human populations is heritable genetic effects resulting from mutations induced in germinal cells. For high dose-rate exposure, the induced mutation rate per gamete generally falls in the range of 10−4 to 10−5 per cGy. The rates per locus are in the range of 10−7 to 10−8 per cGy. Protraction of exposure appears to decrease the mutation rate in rodent systems by a factor of 2 or greater. When all of the experimental data for the various genetic end points are considered, the genetic doubling dose (radiation dose necessary to double the spontaneous mutation rate) for low dose-rate exposure appears to be in the range of 100 cGy.
Chromosomal aberrations
Radiation can induce two types of chromosomal aberrations in mammalian cells. The first have been termed “unstable” aberrations in that they are usually lethal to dividing cells. They include such changes as dicentrics, ring chromosomes, large deletions, and fragments. The frequency of such aberrations correlates well with the cytotoxic effects of radiation.
The second type has been termed “stable” aberrations. These include changes such as small deletions, reciprocal translocations, and aneuploidy changes that do not preclude the cell from dividing and proliferating. Radiation-induced reciprocal translocations may be passed on through many generations of cell replication and emerge in clonal cell populations.17, 18
Such deletions and translocations can result in gene mutations. Specific chromosomal abnormalities are associated with specific tumor types such as the chromosome 8:14 translocation in Burkitt lymphoma. The chromosomal change can result in the activation of a specific oncogene such as a chromosome 13q14 deletion in retinoblastoma (RB).
Radiation-induced genomic instability
Radiation exposure can induce a type of transmissible genetic instability in individual cells that is transmitted to their progeny, leading to a persistent enhancement in the rate at which genetic changes arise in the descendants of the irradiated cell after many generations of replication. This is a nontargeted effect of radiation. The end points include malignant transformation, specific gene mutations, and chromosomal aberrations.
Early evidence was derived from an examination of the kinetics of radiation-induced malignant transformation of cells in vitro.19, 20 Transformed foci did not arise from a single, radiation-damaged cell. Rather, radiation appeared to induce a type of instability in 20–30% of the irradiated cell population; this instability enhanced the probability of the occurrence of a second, neoplastic transforming event, which is rare occurring with the frequency of ∼10−6. This second event occurs with a constant frequency per cell per generation and has the characteristics of a mutagenic event.21
This phenomenon was subsequently demonstrated in a number of experiment systems for various genetic end points.22–25 In terms of mutagenesis, ∼10% of clonal populations derived from single cells surviving radiation exposure showed a significant elevation in the frequency of spontaneously arising mutations compared with clonal populations derived from nonirradiated cells.26, 27 This increased mutation rate persisted for ∼30 to 50 generations postirradiation. An enhancement of both minisatellite28 and microsatellite29 instability has also been observed in the progeny of irradiated cells selected for mutations at the thymidine kinase locus.
Transmission of chromosomal instability has been shown to occur in vivo,30, 31 but susceptibility to radiation-induced chromosomal instability differed significantly among different strains of mice.32, 33 A persistent increase in the rate of cell death has been shown to occur in cell populations many generations after radiation exposure.34–36 Delayed reproductive failure has been linked to chromosomal instability37 and malignant transformation,38, 39 and evidence presented to suggest that DNA is at least one of the critical targets in the initiation of this phenomenon.40
A recent novel phenomenon has been identified and coined “the delayed radioprotective effect.” It is manifested by an enhanced resistance to ionizing radiation hours to days following exposure of cells to thiol-containing drugs such as N-acetylcysteine and captopril that have the ability to elevate intracellular antioxidant enzymes such as manganese superoxide dismutase (MnSOD).41, 42 The underlying mechanism of action responsible for this effect is the activation of the redox-sensitive nuclear transcription factor κB (NFκB) by thiol-reducing agents that subsequently results in the elevated transcription of MnSOD. The resulting 10- to 20-fold elevation of intracellular MnSOD facilitates the prevention and removal of radiation-induced oxidative damage and can enhance survival of cells by 10–30%.
Bystander effects in irradiated cell populations
It has long been thought that the cell nucleus is the target for the important biologic effects of radiation. However, recent evidence shows that targeted cytoplasmic radiation is significantly mutagenic. Damage signals may be transmitted from irradiated to nonirradiated cells in the population, leading to biologic effects in cells that received no radiation exposure,43 for example, the “bystander” effect.
Following the exposure of monolayer cultures of cells to very low fluences of α particles, an enhanced frequency of sister chromatid exchanges (SCE) was observed in 20–40% of cells exposed to fluences by which only 1/1000 to 1/100 cells were traversed by an α particle.44 This effect involves the secretion of cytokines or other factors by irradiated cells that leads to an upregulation of oxidative metabolism in bystander cells.45, 46 Examples of a bystander effect are presented in Figure 2. Incubation with conditioned medium from irradiated cells has cytotoxic effects on nonirradiated cells, which may be related to the release of a factor(s) into the medium.47 An enhanced frequency of specific gene mutations48, 49 as well as chromosomal aberrations50, 51 are observed to occur in bystander cells in populations exposed to very low fluences of α particles.
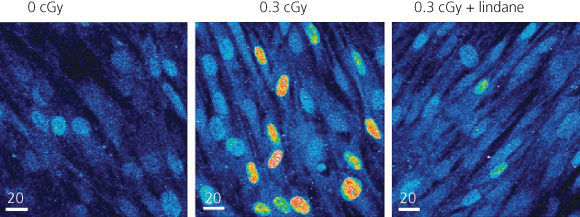
Figure 2 Bystander effect of radiation. The expression of p21Wafl was determined in individual cells in monolayer cultures of human diploid fibroblasts by in situ immunofluorescence. In cultures exposed to 0.3 cGy (<1–2% of nuclei traversed by an α particle), enhanced expression occurred in many nonirradiated cells occurring in clusters (center panel). The effect was suppressed by incubation with lindane, an inhibitor of gap junction-mediated intercellular communication (right panel).
DNA damage in bystander cells appears to differ from that occurring in directly irradiated cells; whereas the mutations induced in directly irradiated cells were primarily partial and total gene deletions, >90% of those arising in bystander cells were point mutations.52 Suggesting that oxidative metabolism is upregulated in bystander cells.45, 46 Bystander effects indicate clearly that damage signals can be transmitted from irradiated to nonirradiated cells.
Adaptive responses
The original description of an adaptive response was made by investigators working with human lymphocytes in which they observed that following exposure to a very low dose of ionizing radiation in the range of 1–10 cGy cells acquired an enhanced resistance to a second but much larger dose of 2 Gy or more.53 The expression of an adaptive response was linked to the requirement of de novo protein synthesis since inhibitors of protein synthesis such as cycloheximide were found to be inhibitory to this inductive effect.54 Adaptive responses can be looked upon as the result of intercellular stress responses. The most studied such response has been identified as being mediated by MnSOD, an antioxidant enzyme localized in the mitochondria of cells, in both normal and malignant cells. As an example, it has been demonstrated that mouse skin JB6P+ epithelial cells exposed to 10 cGy exhibited an enhanced resistance to a subsequent dose of 2 Gy during which time a number of NFκB-regulated genes including MnSOD, p65, phosphorylated extracellular signal-related kinase, cyclin B1, and 14-3-3Z were elevated.55 The importance of elevated MnSOD synthesis in the adaptive response process has been observed in cells following exposure to not only a low dose of ionizing radiation55 but also the cytokine TNFα56 and numerous reductive agents such as amifostine and N-acetylcysteine.42, 57 Treatment of cells with NFκB inhibitors and/or antisense MnSOD oligomers or siRNA MnSOD completely inhibited the adaptive response induced by these agents.55, 56 Both the radiation and cytokine-induced adaptive responses are the result of oxidative damage-initiated processes in contrast to the thiol-induced reductive response in which NFκB is activated following the reduction of cysteine residues on its p50 and p65 subunits,58, 59 a process that can be maintained in a persistent manner with chronic thiol exposure.60
It is now recognized that there are multiple mediators of adaptive responses, each associated with either normal and/or neoplastic cells and different radiation protocols. Under conditions of multifractionated high-dose radiation exposures, each preceded by very low imaging doses, a survivin-mediated adaptive response is expressed in tumor cells.61 Survivin, a member of the inhibitor of apoptosis protein (IAP) group, is primarily found in neoplastic cells and has been identified as an important factor in tumor cell resistance.62, 63 Its overexpression results in elevated tumor cell resistance to radiation- and chemotherapy-induced cell killing and reduced frequencies of apoptosis.64–66 A third adaptive response has been identified in normal cells where lose-dose radiation exposure results in the induction of a metabolic shift from oxidative phosphorylation to aerobic glycolysis in cells and is mediated by the hypoxia-inducible factor HIF-1α.67
DNA damage
Track analysis studies of X-ray interactions in DNA have provided evidence for clustered damage, which results in complex DSB (double-strand break).68 Certain types of DNA base damage such as 8-hydroxydeoxyguanosine and thymine glycols have significant potential biologic importance, but the available data suggest that such isolated base damage by itself probably plays a minor role in radiation mutagenesis. Clustered DNA damage may include abasic sites, oxidized purines, or oxidized pyrimidines.69 The increased efficiency of DNA breaks induced by high-LET radiation appears to result primarily from their greater complexity.70
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


