Chapter Outline
Investigation of membrane defects 229
Osmotic fragility as measured by lysis in hypotonic saline 229
Osmotic fragility after incubating the blood at 37 °C for 24 H 230
Flow cytometric (dye-binding) test 233
Glycerol lysis-time tests 234
Acidified glycerol lysis-time test 234
Cryohaemolysis test 235
Autohaemolysis: spontaneous haemolysis developing in blood incubated at 37 °C for 48 H 235
Membrane protein analysis 237
Detection of enzyme deficiencies in hereditary haemolytic anaemias 237
Screening tests for G6PD deficiency and other defects of the pentose phosphate pathway 237
Fluorescence screening test for G6PD deficiency 238
Methaemoglobin reduction test 239
Detection of heterozygotes for G6PD deficiency 240
Cytochemical tests for demonstrating defects of red cell metabolism 241
Demonstration of G6PD-deficient cells 241
Pyrimidine-5′-nucleotidase screening test 241
Red cell enzyme assays 242
Pyruvate kinase assay 246
Estimation of reduced glutathione 247
Glutathione stability test 248
2,3-Diphosphoglycerate 249
Measurement of red cell 2,3-diphosphoglycerate 249
Oxygen dissociation curve 251
Determining the oxygen dissociation curve 251
The various initial steps to be taken in the investigation of a patient suspected of having a haemolytic anaemia are outlined in Chapter 11 and the changes in red cell morphology that may be found in haemolytic anaemias are illustrated in Chapter 5 . This chapter describes procedures useful in investigating haemolytic anaemias suspected to result from defects within the red cell membrane or deficiency of enzymes important in red cell metabolism.
The precise identification of an enzyme defect is beyond the scope of most haematology laboratories; it may require the isolation and purification of the enzyme and the determination of its kinetic and structural properties. In a service laboratory, it is sufficient to identify the general nature of the defect, whether it be in the membrane or the metabolic pathways of the red cell. In the case of putative metabolic defects, an attempt should be made, where possible, to pinpoint the enzyme involved. The first part of this chapter describes screening tests for spherocytosis, including hereditary spherocytosis (HS) and for glucose-6-phosphate dehydrogenase (G6PD) deficiency. The later sections of the chapter describe specific enzyme assays and the measurement of 2,3-diphosphoglycerate (2,3-DPG) (also known as 2,3-bisphosphoglycerate, 2,3-BPG) and reduced glutathione (GSH).
Most of the enzyme assays have been standardised by the International Council for Standardisation in Haematology (ICSH). Commercial kits are also available for some quantitative assays and screening tests. These are noted in the relevant sections.
Investigation of membrane defects
The osmotic fragility test gives an indication of the surface area/volume ratio of erythrocytes. Its greatest usefulness is in the diagnosis of HS. The test may also be used in screening for thalassaemia. Red cells that are spherocytic, for whatever cause, take up less water in a hypotonic solution before rupturing than do normal red cells.
Other tests that demonstrate red cell membrane defects include glycerol lysis time, cryohaemolysis, autohaemolysis and, more specifically, membrane protein analysis.
Procedures to assess red cell flexibility (rigidity) using polycarbonate membrane filtration, and red cell deformability measurements on specialised equipment such as the Laser-assisted Optical Rotational Cell Analyser (Lorrca) have been described elsewhere.
Osmotic fragility as measured by lysis in hypotonic saline
Principle
The method to be described is based on that of Parpart et al . Small volumes of blood are mixed with a large excess of buffered saline solutions of varying concentration. The fraction of red cells lysed at each saline concentration is determined colorimetrically. The test is normally carried out at room temperature (15–25 °C).
Reagents
Prepare a stock solution of buffered sodium chloride, osmotically equivalent to 100 g/l (1.71 mol/l) NaCl, as follows: dissolve NaCl, 90 g; Na 2 HPO 4 , 13.65 g (or Na 2 HPO 4 2H 2 O, 17.115 g); and NaH 2 PO 4 .2H 2 O, 2.34 g in water. Adjust the final volume to 1 litre. This solution will keep for months at 4 °C in a well-stoppered bottle. Salt crystals may form on storage and must be thoroughly redissolved before use.
In preparing hypotonic solutions for use, it is convenient to make first a 10 g/l solution from the 100 g/l NaCl stock solution by dilution with water. Dilutions equivalent to 9.0, 7.5, 6.5, 6.0, 5.5, 5.0, 4.0, 3.5, 3.0, 2.0 and 1.0 g/l are convenient concentrations. Intermediate concentrations such as 4.75 and 5.25 g/l are useful in critical work and an additional 12.0 g/l dilution should be used for incubated samples.
It is convenient to make up 50 ml of each dilution. The solutions keep well at 4 °C if sterile, but should be inspected for moulds before use and discarded if moulds develop.
Method
Heparinised venous blood or defibrinated blood may be used; oxalated or citrated blood is not suitable because of the additional salts added to it. The test should be carried out within 2 h of collection with blood stored at room temperature or within 6 h if the blood has been kept at 4 °C.
- 1.
Deliver 5.0 ml of each of the 11 saline solutions into 12 × 75 mm test tubes. Add 5.0 ml of water to the 12th tube.
- 2.
Add to each tube 50 μl of well-mixed blood and mix immediately by inverting the tubes several times, avoiding foam.
- 3.
Leave the suspensions for 30 min at room temperature. Mix again and then centrifuge for 5 min at 1200 g .
- 4.
Remove the supernatants and estimate the amount of lysis in each using a spectrometer at a wavelength setting of 540 nm or a photoelectric colorimeter provided with a yellow–green (e.g. Ilford 625) filter. Use as a blank the supernatant from tube 1 (osmotically equivalent to 9 g/l NaCl).
- 5.
Assign a value of 100% lysis to the reading with the supernatant of tube 12 (water) and express the readings from the other tubes as a percentage of the value of tube 12. Plot the results against the NaCl concentration ( Fig. 12-1 ).
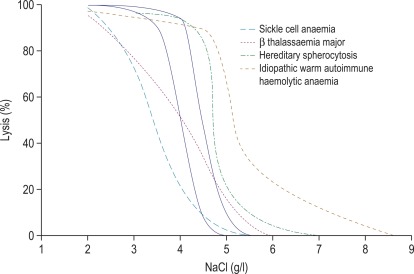
Figure 12-1
Osmotic fragility curves.
Osmotic fragility curves of patients suffering from the following: sickle cell anaemia, β thalassaemia major, hereditary spherocytosis and ‘idiopathic’ warm autoimmune haemolytic anaemia. The normal range is indicated by the unbroken lines.
Notes
- 1.
The measurement of osmotic fragility is a simple procedure that requires a minimum of equipment. It will yield gratifying results if carried out carefully.
- 2.
The blood must be delivered into the 12 tubes with great care. The critical point is not that the amount be exactly 50 μl, but rather that the amount added to each tube must be the same. Two methods are recommended:
- a.
Using an automatic pipette, after aspirating the blood gently, wipe the outside with tissue paper, taking care not to suck out any blood from the inside of the tip by capillary action. The blood is then delivered into the saline solution and the pipette is rinsed in and out several times until no blood is visible inside its tip.
The tip has to be changed before moving on to the next tube. This procedure takes time and may result in an increased exposure for the first few tubes. It is therefore advisable to start the timing only after the addition of the sample to the first tube.
- b.
Using a Pasteur pipette with a perfectly flat end, 1 mm in diameter, suck up about 1 ml of blood, avoiding any bubbles and wipe the outside of the pipette. With the pipette held vertically above tube 1, deliver a single drop (about 50 μl) without the blood touching the wall of the tube. Then deliver single drops into the remaining 11 tubes.
Method (b) appears to be primitive, but with practice it is perfectly satisfactory; it is also more economical and much faster than method (a). With either method, the best way to test its accuracy is to do a preliminary test by delivering the blood into several tubes all containing the same saline solution (e.g. either 3.0 or 1.0 g/l). The readings with the supernatants should be all within 5% of each other.
- 3.
If the amount of blood available is limited (e.g. from babies) and the spectrometer takes 1 ml cuvettes, the volumes can be scaled down to 1 ml of saline solution and 10 μl of blood. However, to deliver 10 μl of blood reproducibly is not easy. With method (b), a Pasteur pipette or capillary pipette with a much smaller diameter, calibrated to give 10 μl drops of blood, would have to be used. It is then more difficult to maintain accuracy. Method (a) may be preferable in this case.
- 4.
With the method using 50 μl of blood and with nonanaemic blood, the reading for 100% lysis will be about 0.7. With a modern spectrometer, any figure between 0.5 and 1.5 is acceptable. If the value is lower than 0.5, the test should be repeated using more blood or less saline (the reverse if the reading is higher than 1.5). With photoelectric colorimeters, values higher than 0.5 are often not very accurate.
- 5.
When transferring the supernatant from a tube to the spectrometer cuvette, care has to be taken not to disturb the pellet. If it is well packed, the supernatant can simply be poured from the tube into the cuvette; with a spectrometer provided with an automatic suction device, this is usually satisfactory. Alternatively, a plastic Pasteur pipette can be used.
- 6.
Even when a normal range has been established, it is essential always to run a normal control sample along with that of the patients to be tested to check, for example, the saline solutions.
- a.
The sigmoid shape of the normal osmotic fragility curve indicates that normal red cells vary in their resistance to hypotonic solutions. Indeed, this resistance varies gradually (osmotically) as a function of red cell age, with the youngest cells being the most resistant and the oldest cells being the most fragile. The reason for this is that old cells have a higher sodium content and a decreased capacity to pump out sodium.
Osmotic fragility after incubating the blood at 37 °C for 24 H
Method
Defibrinated blood should be used, care being taken to ensure that sterility is maintained.
Incubate 1 ml or 2 ml volumes of blood in sterile 5 ml bottles. It is advisable to set up the samples in duplicate in case one has become infected, as indicated by gross lysis and change in colour.
After 24 h, if no infection is evident, pool the contents of the duplicate bottles after thoroughly mixing the sedimented red cells in the overlying serum and estimate the fragility as previously described.
Because the fragility may be markedly increased ( Fig. 12-2 ), set up additional hypotonic solutions containing 7.0 g/l and 8.0 g/l NaCl. In addition, use a solution equivalent to 12.0 g/l NaCl because sometimes, as in HS, lysis may take place in 9.0 g/l NaCl. In this case, use the supernatant of the tube containing 12.0 g/l NaCl as the blank in the colorimetric estimation.

The incubation fragility test is conveniently combined with the estimation of the amount of spontaneous autohaemolysis (p. 235).
Factors affecting osmotic fragility tests
In carrying out osmotic fragility tests by any method, three variables capable of markedly affecting the results must be controlled, quite apart from the accuracy with which the saline solutions have been made up. These are as follows:
- 1.
The relative volumes of blood and saline
- 2.
The final pH of the blood in saline suspension
- 3.
The temperature at which the tests are carried out.
A proportion of 1 volume of blood to 100 volumes of saline is chosen because the concentration of blood is so small that the effect of the plasma on the final tonicity of the suspension is negligible. When weak suspensions of blood in saline are used, it is necessary to control the pH of the hypotonic solutions and it is for this reason that phosphate buffer is added to the saline. Even so, small differences will be found between the fragility of venous blood and maximally aerated (i.e. oxygenated) blood. For the most accurate results, it is recommended that the blood is mixed until bright red. Finally, it is ideal for tests to be carried out always at the same temperature, although for most purposes room temperature is sufficiently constant.
The extent of the effect of pH and temperature on osmotic fragility was well illustrated in the paper of Parpart et al . The effect of pH is more important: a shift of 0.1 of a pH unit is equivalent to altering the saline concentration by 0.1 g/l, the fragility of the red cells being increased by a decrease in pH. An increase in temperature decreases the fragility, an increase of 5 °C being equivalent to an increase in saline concentration of about 0.1 g/l.
Lysis is virtually complete at the end of 30 min at 20 °C and the hypotonic solutions may be centrifuged at the end of this time.
Further details of the factors that affect and control haemolysis of red cells in hypotonic solutions were given by Murphy.
Recording the results of osmotic fragility tests
In the past, osmotic fragility most often has been expressed in terms of the highest concentration of saline at which lysis is just detectable (initial lysis or minimum resistance) and the highest concentration of saline in which lysis appears to be complete (complete lysis or maximum resistance). It is, however, useful also to record the concentration of saline causing 50% lysis (i.e. the median corpuscular fragility, MCF) and to inspect the entire fragility curve ( Fig. 12-1 ). The findings in health are summarised in Table 12-1 .
| Fresh Blood (g/l NaCl) | Blood Incubated 24 h, 37 °C (g/l NaCl) | |
|---|---|---|
| Initial lysis | 5.0 | 7.0 |
| Complete lysis | 3.0 | 2.0 |
| MCF (50% lysis) | 4.0–4.45 | 4.65–5.9 |
Alternative methods of recording osmotic fragility.
Two simple alternative methods of recording the results quantitatively are available: the data may be plotted on probability paper or increment-haemolysis curves can be drawn. Both methods emphasise heterogeneity of the cell population with respect to osmotic fragility. If the observed amounts of lysis of normal blood are plotted on the probability scale against concentrations of saline, an almost straight line can be drawn through the points; the line is only skewed where lysis is almost complete. This method enables the MCF to be read off with ease.
In disease, tailed curves also skew the line to varying degrees at the other end of the probability plot. To obtain increment-haemolysis curves, the differences in lysis between adjacent tubes are plotted against the corresponding saline concentrations. Definitely bimodal curves may be obtained during recovery from a haemolytic episode.
Interpretation of results
The osmotic fragility of freshly taken red cells reflects their ability to take up a certain amount of water before lysing. This is determined by their volume-to-surface area ratio. The ability of the normal red cell to withstand hypotonicity results from its biconcave shape, which allows the cell to increase its volume by about 70% before the surface membrane is stretched; once this limit is reached lysis occurs. Spherocytes have an increased volume-to-surface area ratio; their ability to take in water before stretching the surface membrane is thus more limited than normal, and they are therefore particularly susceptible to osmotic lysis. The increase in osmotic fragility is a property of the spheroidal shape of the cell and is independent of the cause of the spherocytosis. Characteristically, osmotic fragility curves from patients with HS who have not been splenectomised show a ‘tail’ of very fragile cells ( Fig. 12-3 ). When plotted on probability paper, the graph indicates two populations of cells: the very fragile and the normal or slightly fragile. After splenectomy the red cells are more homogeneous, the osmotic fragility curve indicating a more continuous spectrum of cells, from fragile to normal.

Decreased osmotic fragility indicates the presence of unusually flattened red cells (leptocytes) in which the volume-to-surface area ratio is decreased. Such a change occurs in iron deficiency anaemia and thalassaemia in which the red cells with a low mean cell haemoglobin (MCH) and mean cell volume (MCV) are unusually resistant to osmotic lysis ( Fig. 12-1 ). A simple one-tube osmotic fragility is a useful screening test for β thalassaemia and some haemoglobinopathies in countries with a high incidence of these abnormalities (p. 556). Reticulocytes and red cells from patients who have been splenectomised also tend to have a greater amount of membrane compared with normal cells and are osmotically resistant. In liver disease, target cells may be produced by passive accumulation of lipid, and these cells, too, are resistant to osmotic lysis.
The osmotic fragility of red cells after incubation for 24 h at 37 °C is also a reflection of their volume-to-surface area ratio, but the factors that alter this ratio are more complicated than in fresh red cells. The increased osmotic fragility of normal red cells, which occurs after incubation ( Fig. 12-2 ), is mainly caused by swelling of the cells associated with an accumulation of sodium that exceeds loss of potassium. Such cation exchange is determined by the membrane properties of the red cell, which control the passive flux of ions, and the metabolic competence of the cell, which determines the active pumping of cations against concentration gradients. During incubation for 24 h, the metabolism of the red cell becomes stressed and the pumping mechanisms tend to fail, one factor being a relative lack of glucose in the medium.
The osmotic fragility of red cells that have an abnormal membrane, such as those of HS and hereditary elliptocytosis (HE), increases abnormally after incubation ( Fig. 12-2 ). Similar results occur in hereditary stomatocytosis. The results with red cells with a glycolytic deficiency, such as those of pyruvate kinase (PK) deficiency, are variable. In severe deficiencies, osmotic fragility may increase substantially ( Fig. 12-2 ), but in other cases, the fragility may decrease owing to a greater loss of potassium than gain of sodium. In thalassaemia major and minor, osmotic fragility is frequently markedly reduced after incubation, again owing to a marked loss of potassium. A similar, although usually less marked, change is seen in iron deficiency anaemia.
To summarise, measurement of red cell osmotic fragility provides a useful indication as to whether a patient’s red cells are normal because an abnormal result invariably indicates abnormality. The reverse is, however, not true (i.e. a result that is within the normal range does not mean that the red cells are normal). The findings in some important haemolytic anaemias are summarised in Table 12-2 .
| Condition | Notes |
|---|---|
| A. Associated with Increased Osmotic Fragility (OF) | |
| Hereditary spherocytosis (HS) | Entire curve may be ‘shifted to the right’, or most of it may be within the normal range but with a ‘tail’ of fragile cells. Curve within normal range in 10–20% of cases. After incubation for 24 h, abnormalities usually more marked, but still some false-negative results. Splenectomy does not affect MCF but reduces the tail of fragile cells |
| Hereditary elliptocytosis (HE) | As in HS, but in general changes less marked. Abnormal OF usually correlates with severity of haemolysis (i.e. OF is normal in nonhaemolytic HE) |
| Hereditary stomatocytosis | As in HS with large osmotically fragile cells with low MCHC |
| Other inherited membrane abnormalities | Results variable; with milder disorders curve more likely to be abnormal after incubation for 24 h |
| Autoimmune haemolytic anaemia | Tail of fragile cells roughly proportional to number of spherocytes; rest of curve normal (or even left-shifted on account of reticulocytosis) |
| B. Associated with Decreased OF | |
| Thalassaemia | MCF decreased in all forms of thalassaemia, except in some α thalassaemia heterozygotes; usually the entire curve is left-shifted |
| Enzyme abnormalities | OF usually normal (anaemia originally referred to as hereditary nonspherocytic), but tail of highly resistant cells may be seen on account of high reticulocyte count. After incubation for 24 h, there may be a tail of fragile cells |
| Hereditary xerocytosis | Increased resistance to osmotic lysis and increased MCHC |
| Iron deficiency | Curve shifted to left, wholly or partly, depending on proportion of hypochromic red cells |
Flow cytometric (dye-binding) test
Principle
The osmotic fragility test lacks specificity and sensitivity and may fail to detect atypical or mild HS. Moreover, it can be affected by factors unrelated to red cell cytoskeletal defects; for example, positive results may be obtained for red cells from patients who are pregnant or who have immune or other haemolytic anaemias or renal failure. The flow cytometric (dye-binding) test of King and colleagues measures the fluorescence intensity of intact red cells labelled with eosin-5-maleimide (EMA), which reacts covalently with Lys-430 on the first extracellular loop of Band 3 protein. The N-terminal cytoplasmic domain of Band 3 interacts with ankyrin and protein 4.2, which in turn crosslink with the spectrin-based cytoskeleton and stabilises the membrane lipid bilayer. Deficiency or abnormality of Band 3 may result in decreased fluorescence. This is seen in HS red cells but has also been observed in cases of hereditary pyropoikilocytosis (HPP), Southeast Asian ovalocytosis, congenital dyserythropoietic anaemia Type II and the stomatocytic variant, cryohydrocytosis. Blood samples in ethylenediaminetetra-acetic acid (EDTA) can be analysed for up to 48 h after collection provided they have been stored in the refrigerator.
Reagents
Eosin-5-maleimide (EMA)
EMA is light sensitive and must be kept in the dark, preferably wrapped in aluminium foil and stored at 4 °C. Prepare a stock solution by dissolving 1 mg in 1 ml of phosphate buffered saline (PBS). Mix well and store in 200 μl aliquots at − 20 °C.
Bovine serum albumin (30%) solution (BSA)
Available commercially. Dilute to 0.5% with PBS.
Phosphate buffered saline
Tablets are available commercially for dissolving in water (e.g. Oxoid Dulbecco ‘A’ tablets, www.oxoid.com/uk ). Alternatively, prepare by adding equal volumes of iso-osmotic phosphate buffer and 9 g/l NaCl:
Phosphate buffer, iso-osmotic, pH 7.2:
- (A)
NaH 2 PO 4 .2H 2 O (150 mmol/l) − 23.4 g/l
- (B)
Na 2 HPO 4 (150 mmol/l) − 21.3 g/l.
Add 24 ml reagent A to 76 ml reagent B.
- (A)
Method
Thaw a tube of stock EMA solution in the dark at room temperature and dilute with an equal volume of PBS to obtain a working solution of 0.5 mg/ml. Mix 5 μl of washed packed red cells with 25 μl of EMA working solution in a plastic microfuge tube. Set up control tubes from blood of normal individuals, and perform all tests in duplicate. Leave a rack with the tubes in a cupboard in the dark at room temperature for 30 min. Mix and return to the cupboard for a further 30 min.
Then, spin the tubes in a bench-top microfuge for 5–10 s and remove the supernatant dye carefully with a fine-tip pipette.
Wash the labelled red cells three times with 500 μl PBS containing 0.5% BSA. The third wash should be colourless. If it is still pink, suggesting that traces of dye particle remain in the tube, discard the sample and repeat the cell-labelling procedure.
Resuspend the packed red cells in 500 μl of the PBS–BSA wash solution. Transfer 100 μl of the cell suspension into a plastic flow cytometer tube and add 1.4 ml of wash solution. Keep the cell suspensions in the dark by wrapping in aluminium foil until use. Set the analyser thresholds (gate) for red blood cells and count each sample for a minimum of 15 000 events. Select the FITC (fluorescein isothiocyanate) channel and record the mean fluorescence intensity (MFI). Compare the test with the mean value of several control samples analysed at the same time.
Interpretation of results
Results are expressed as a ratio of mean value of the test to control samples. Each laboratory should set the reference range and cut-off values for its own instrument. In our laboratory, a range of 0.83 to 1.17 is considered normal. HS red cells give a ratio of less than 0.8. Values as low as 0.6 may be seen in classic HS. Similar or even lower values are seen in the much rarer HPP, but the latter is readily distinguished from HS on the basis of red cell morphology and a very low MCV. A reduced ratio may also be encountered in cases of Southeast Asian ovalocytosis, congenital dyserythropoietic anaemia Type II and cryohydrocytosis, but these are associated with different types of red cell atypia and may generally be ruled out by examination of a stained blood film. EMA binding has a high sensitivity in the diagnosis of HS, however in a very small number of cases of typical HS the MFI ratio is normal. If HS is suspected and EMA binding normal, a second screening test may be informative.
Glycerol lysis-time tests
The osmotic fragility test is somewhat cumbersome and requires 2 ml or more of whole blood. It is thus not suitable for use in newborn babies or as a population screening test. In 1974, Gottfried and Robertson introduced a glycerol lysis-time (GLT) test, a one-tube test to measure the time taken for 50% haemolysis of a blood sample in a buffered hypotonic saline–glycerol mixture. The original method had greater sensitivity in the osmotic-resistant range, but it also could identify most patients with HS by a shorter GLT 50 . Better differentiation of HS blood from normal was obtained by 24 h incubation of samples and by modifying the glycerol reagent. Zanella et al . modified the original test further by decreasing the pH. There is some loss of specificity for HS with the acidified glycerol lysis-time test (AGLT) compared with the original method, but in practice this loss is unimportant.
Acidified glycerol lysis-time test
Principle
Glycerol present in a hypotonic buffered saline solution slows the rate of entry of water molecules into the red cells so that the time taken for lysis may be more conveniently measured. Like the osmotic fragility test, differentiation can be made between spherocytes and normal red cells.
Reagents
Phosphate buffered saline
Add 9 volumes of 9.0 g/l (154 mmol/l) NaCl to 1 volume of 100 mmol/l phosphate buffer (2 volumes of 14.9 g/l Na 2 HPO 4 added to 1 volume of 13.61 g/l KH 2 PO 4 ). Adjust the pH to 6.85 ± 0.05 at room temperature (15–25 °C). This adjustment must be accurate.
Glycerol reagent
300 mmol/l. Add 23 ml of glycerol (27.65 g AR grade) to 300 ml of PBS and bring the final volume to 1 litre with water.
Method
Add 20 μl of whole blood, anticoagulated with EDTA, to 5.0 ml of PBS, pH 6.85. Mix the suspension carefully.
Transfer 1.0 ml to a standard 4 ml cuvette of a spectrometer equipped with a linear-logarithmic recorder. Fix the wavelength at 625 nm and start the recorder. Add 2.0 ml of the glycerol reagent rapidly to the cuvette with a 2 ml syringe or automatic pipette and mix well.
The rate of haemolysis is measured by the rate of fall of turbidity of the reaction mixture. The results are expressed as the time required for the optical density to fall to half the initial value (AGLT 50 ). The test can also be carried out using a colorimeter and stopwatch.
Results
Normal blood takes more than 30 min (1800 s) to reach the AGLT 50 . The time taken is similar for blood from normal adults, newborn infants and cord samples. In patients with HS, the range of the AGLT 50 is 25–150 s. A short AGLT 50 may also be found in chronic renal failure, chronic leukaemias and autoimmune haemolytic anaemia; it also may be found in some pregnant women.
Significance of the acidified glycerol lysis-time test
The same principles apply as with the osmotic fragility test. Cells with a high volume-to-surface area ratio resist swelling for a shorter time than normal cells. This applies to all spherocytes, whether the spherocytosis is caused by HS or other mechanisms. The test is particularly useful in screening family members of patients with HS where morphological changes are too minor to indicate clearly whether the disorder is present.
Cryohaemolysis test
Principle
Whereas osmotic fragility may be abnormal in any condition where spherocytes occur, it has been suggested that cryohaemolysis is specific for HS. This appears to result from the fact that the latter is dependent on factors that are related to molecular defects of the red cell membrane rather than to changes in the surface area-to-volume ratio. The test can be carried out on EDTA blood up to 1 day old.
Reagent
Buffered 0.7 mol/l sucrose
Dissolve 23.96 g sucrose in 100 ml of 50 mmol/l phosphate buffer, pH 7.4. This can be stored frozen in 2 ml aliquots in tubes ready for use.
Method
- 1.
Centrifuge the blood and wash the red cells three times with cold (4 °C) 9 g/l NaCl. Make a suspension of 50–70% cells in the saline and keep on ice until tested.
- 2.
Prepare 2 ml volumes of reagent, thawing if frozen, and stand for 10 min in a 37 °C water bath to equilibrate.
- 3.
Pipette 50 μl of the cell suspension into each of 2 tubes of the warmed reagent, vortex immediately for a few seconds and then incubate for exactly 10 min at 37 °C.
- 4.
Without delay, transfer the tubes to an ice bath for another 10 min, vortex for a few seconds and then centrifuge to sediment the remaining cells. Transfer some of the supernatant to a clean tube.
- 5.
Prepare a 100% haemolysate solution by pipetting 50 μl of the original sample into 2 ml of water. Centrifuge and dilute 200 μl of the supernatant in 4 ml of water.
- 6.
Read absorbance at 540 nm of the test and the 100% lysis samples.
% c r y o h a e m o l y s i s = A 540 test A 540 haemolysate × 21 × 100
Interpretation
Streichman et al . reported the range of cryohaemolysis in normal subjects to be 3–15%, whereas in HS there was > 20% lysis. However, it is recommended that individual laboratories establish their own reference values for the method. We have found that most normal samples give < 3% lysis. Increased lysis is not exclusive to HS and may be observed in hereditary stomatocytosis.
Autohaemolysis: spontaneous haemolysis developing in blood incubated at 37 °C for 48 H
The autohaemolysis test is useful as an initial screen in suspected cases of haemolytic anaemia. It provides information about the metabolic competence of the red cells and helps to distinguish membrane and enzyme defects if the results of the tests are taken together with other observations such as morphology, inheritance and the presence or absence of associated clinical disorders.
Principle
Aliquots of blood are incubated both with and without sterile glucose solution at 37 °C for 48 h. After this period, the amount of spontaneous haemolysis is measured colorimetrically.
Method
It is essential to use aseptic techniques in setting up the autohaemolysis test to maintain sterility throughout the incubation period.
Use blood collected into ACD or defibrinated blood (see previous editions). Deliver four 1 ml or 2 ml samples into sterile 5 ml capped bottles. Retain a portion of the original sample; separate and store this as the preincubation serum.
Add to two of the bottles 50 or 100 μl of sterile 100 g/l glucose solution, so as to provide a concentration of glucose in the blood of at least 30 mmol/l. Make sure that the caps of the bottles are tightly closed and place the series of bottles in the incubator at 37 °C. A sample from a known normal individual should be run in parallel as a control.
After 24 h, thoroughly mix the content by gentle swirling. After incubating for a further 24 h, inspect the samples for signs of infection, thoroughly mix again, then from each bottle remove a sample for the estimation of the packed cell volume (PCV) (by the microhaematocrit method) and haemoglobin concentration (Hb) and centrifuge the remainder to obtain the supernatant serum.
Estimate the spontaneous lysis by means of a colorimeter or a spectrometer at 540 nm.
As a rule, it is convenient to make a 1 in 10 dilution of the incubated serum in cyanide-ferricyanide (Drabkin) solution (p. 20), unless there is marked haemolysis, when a 1 in 25 or 1 in 50 dilution is more suitable. A corresponding dilution of the preincubation serum is used as a blank and a 1 in 100 or 1 in 200 dilution of the whole blood in Drabkin solution indicates the total amount of Hb present and serves as a standard.
Calculate the percentage lysis, allowing for the change in PCV resulting from the incubation as follows:
Lysis % = R t − B R 0 × D 0 D t × 1 − PCV t × 100
The reading at time t is multiplied by (1 − PCV t ) so as to give the concentration that would be found if the liberated haemoglobin were dissolved in whole blood (i.e. in both plasma and red cell compartments), not in the plasma compartment alone.
Normal range of autohaemolysis
Lysis at 48 h: without added glucose, 0.2% to 2.0%; with added glucose, 0% to 0.9%.
The results obtained are sensitive to slight differences in technique and each laboratory should use a carefully standardised procedure and establish its own normal range. If the amount of liberated haemoglobin is small, it is more accurate (although more time consuming) to measure lysis by a chemical method rather than by a direct spectrometric method (p. 216). It can also be measured directly by a simple and rapid procedure with a HemoCue Plasma/Low Hb system ( www.hemocue.com ).
Significance of increased autohaemolysis
Little or no lysis takes place when normal blood is incubated for 24 h under sterile conditions and the amount present after 48 h is small. If glucose is added so that it is present throughout the incubation, the development of lysis is markedly slowed. The amount of autohaemolysis that occurs after 48 h with and without glucose is determined by the properties of the membrane and the metabolic competence of the red cell. In membrane disorders such as HS, the rate of glucose consumption is increased to compensate for an increased cation leak through the membrane. During the 48 h incubation, glucose is therefore used up relatively rapidly so that energy production fails more quickly than normal unless glucose is added. This is one factor that contributes to the increased rate of autohaemolysis in HS. Usually, but not always, the addition of glucose to the blood decreases the rate of autohaemolysis in HS. This was referred to as Type 1 autohaemolysis. When the utilisation of glucose via the glycolytic pathway is impaired, as in PK deficiency, the rate of autohaemolysis at 48 h is usually increased but glucose fails to correct or may even aggravate lysis (Type 2 autohaemolysis). Although a similar result may be seen in severe HS (Type B), in the absence of spherocytosis, failure of glucose to diminish autohaemolysis is a strong indication of a glycolytic block. Blood from patients with G6PD deficiency or other disorders of the pentose phosphate pathway may undergo a slight increase in autohaemolysis (without additional glucose), which is corrected by the addition of glucose. Commonly, the result is normal, but examination of the incubated blood may show an increase in methaemoglobin (Hi) (discussed later). Not all glycolytic enzyme deficiencies give a Type 2 reaction so that a Type 1 result does not exclude the possibility of such a defect.
In the acquired haemolytic anaemias, the results of the autohaemolysis test are variable and generally not very helpful in diagnosis. In the autoimmune haemolytic anaemias, lysis may be increased in the absence of additional glucose but the effect of added glucose is unpredictable. In paroxysmal nocturnal haemoglobinuria (PNH), the autohaemolysis of aerated defibrinated blood is usually normal.
Autohaemolysis may be increased in haemolytic anaemias caused by oxidant drugs or when there are defects in the reducing power of the red cell. Heinz bodies, Hi or both will be detectable at the end of incubation. Normally, red cells produce < 4% Hi after 48 h incubation and Heinz bodies are not seen. Red cells containing an unstable haemoglobin also contain Heinz bodies at the end of the incubation period and increased amounts of Hi.
The nucleosides adenosine, guanosine and inosine, like glucose, diminish the rate of autohaemolysis when added to blood. Remarkably, adenosine triphosphate (ATP) strikingly retards haemolysis in PK deficiency, although glucose itself is ineffective. ATP does not pass the red cell membrane.
The autohaemolysis test lacks specificity. This has drawn much criticism on the test, including the suggestion that it has no place in the screening of blood for inherited defects. The best way to detect metabolic defects in red cells is undoubtedly to measure glucose consumption, lactate production and the contribution to metabolism of the pentose phosphate pathway. These measurements are, unfortunately, difficult and are likely to be undertaken only by specialised laboratories. The autohaemolysis test does provide some information about the metabolic competence of the red cells and helps to distinguish membrane defects from enzyme defects.
In summary, we feel that the autohaemolysis test is still useful in the investigation of patients who have or who may have chronic haemolytic anaemia for the following reasons:
- 1.
If the result is entirely normal, an intrinsic red cell abnormality is unlikely.
- 2.
If abnormal haemolysis is fully corrected by glucose, a metabolic abnormality is unlikely and a membrane abnormality is likely.
- 3.
If abnormal haemolysis shows little or no correction by glucose, a metabolic abnormality is likely, provided that obvious features of spherocytosis are not present on the blood film.
Thus, in our experience, a combination of red cell morphology with the results of the autohaemolysis tests makes it possible to differentiate membrane abnormalities from enzyme deficiencies in the vast majority of cases.
Membrane protein analysis
Defects of red cell membrane proteins that constitute the cytoskeleton are associated with congenital haemolytic anaemias accompanied by characteristic morphological features. Their analysis is generally only possible in the setting of a reference laboratory. Sodium dodecyl sulphate – polyacrylamide gel electrophoresis (SDS-PAGE) of the membranes will identify qualitative and quantitative alterations in the specific proteins. Densitometry of protein bands on the gel gives an overall profile showing spectrin, ankyrin, Band 3 (the anion transport protein) and protein 4.2. Spectrin variants may be detected after limited trypsin digestion of spectrin extracted from the red cell membranes; an increase in spectrin dimer is indicative of an unstable tetramer, leading to susceptibility to red cell fragmentation in HE and HPP.
Quantitative and qualitative defects of cytoskeletal and other membrane proteins found in hereditary haemolytic anaemias are listed in Table 12-3 .
| Band | Protein | Haemolytic Anaemia |
|---|---|---|
| 1 | α spectrin | HE, HS, HPP |
| 2 | β spectrin | HE, HS |
| 2.1 | Ankyrin | HS |
| 3 | Anion exchanger | HS, SAO, CDAII, CHC |
| 4.1 | Protein 4.1 | HE |
| 4.2 | Pallidin (Protein 4.2) | HS |
| 7.2b | Stomatin | OHSt, CHC |
| PAS-1 | Glycophorin A | CDAII |
| PAS-2 | Glycophorin C | HE |
| ND | Rh-associated glycoprotein | OHSt |
| ND | Glucose transporter (Glut 1) | CHC |
| ND | Piezo 1 | DHSt |
| ND | Gardos channel (KCa3.1) | DHSt |
| ND | ABCB6 | FP |
Detection of enzyme deficiencies in hereditary haemolytic anaemias
It is feasible for most haematology laboratories to identify deficiencies of G6PD and PK and to indicate where the probable defect lies in less common disorders. Detailed investigation of the aberrant enzymes and of the metabolism of the abnormal cells is probably best undertaken by specialised laboratories. Comprehensive accounts of methods available for studying red cell metabolism are to be found in Beutler’s Red Cell Metabolism, a Manual of Biochemical Methods and in the ICSH recommendations.
There are two stages in the diagnosis of red cell enzyme defects: first, screening procedures; and second, specific enzyme assays. The simple nonspecific screening procedures such as the osmotic fragility and autohaemolysis tests, which have already been described, may indicate the presence of a metabolic disorder, and simple biochemical tests are available to show whether the disorder is in the pentose phosphate or the Embden–Meyerhof pathways; these intermediate stages of glycolysis are illustrated in Figure 12-4 .
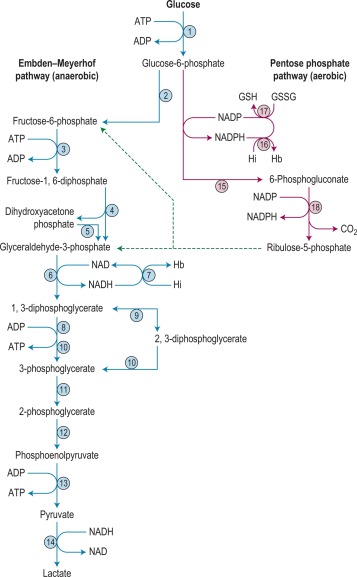
These investigations may be augmented by quantitation of the major red cell metabolites 2,3-DPG, ATP and GSH, which are present at millimolar concentrations and which can be assayed conveniently by spectrometric techniques. Metabolic block in the Embden–Meyerhof pathway is most accurately pinpointed by measurement of the concentration of glycolytic intermediates with demonstration of accumulation of metabolites proximal and depletion of metabolites distal to the defective step ( Fig. 12-4 ). These assays, which are generally confined to specialised laboratories, must be performed on deproteinised red cell extracts immediately after preparation.
Screening tests for G6PD deficiency and other defects of the pentose phosphate pathway
Many variants of the red cell enzyme G6PD have been detected and the methods used to identify variants have been standardised. Inheritance is sex-linked since the enzyme is encoded by a gene on the X chromosome. Variants that have deficient activity produce one of several types of clinical disorders. The two most common variants are the Mediterranean type, which has very low activity and which may lead to favism (i.e. acute intravascular haemolysis following the ingestion of broad beans), and the A − type found in Black populations in West Africa, the USA, the UK and elsewhere, which leads to primaquine sensitivity. Both groups are susceptible to haemolysis produced by oxidant drugs and infections.
Much less frequently, a chronic nonspherocytic haemolytic anaemia is produced by rare variants of the enzyme. Severe neonatal jaundice with anaemia occurs in about 5% of patients who have major deficiencies of enzyme activity.
G6PD deficiency in hemizygous (male) or homozygous (female) individuals may be readily detected by screening tests, but it is more difficult to detect heterozygous (female) carriers. Other defects of the pentose phosphate pathway also lead to deficiency in the reducing power of the red cell. The clinical syndromes associated with these defects include intravascular haemolysis, with or without methaemoglobinaemia, in response to oxidative drugs.
G6PD catalyses the oxidation of glucose-6-phosphate (G6P) to 6-phosphogluconate (6PG) with the simultaneous reduction of nicotinamide adenine dinucleotide phosphate (NADP) to reduced NADP (NADPH):
G 6 P + NADP → G 6 PD 6 PG + NADPH
In a second, consecutive, oxidative reaction, 6PG is converted to 6-phosphogluconolactone, with reduction of a further molecule of NADP to NADPH. The lactone then undergoes decarboxylation to ribulose 5-phosphate through a reaction catalysed by a specific lactonase, but which can also take place spontaneously. Thus the overall reaction catalysed by 6PG dehydrogenase (6PGD) can be written as follows:
6 PG + NADP → 6 PGD Ru 5 P + CO 2 + NADPH
The release of CO 2 drives the reaction to the right so that in practice the pathway is not reversible.
NADPH is an important reducing compound for the conversion of oxidised glutathione (GSSG) to GSH ( Fig. 12-4 ) and under conditions of stress, the reconversion of Hi to haemoglobin. Screening tests for G6PD deficiency depend on the inability of cells from deficient subjects to convert an oxidised substrate to a reduced state. The substrates used may be the natural one of the enzyme, NADP or other naturally occurring substrates linked by secondary reactions to the enzyme, for example, GSSG or Hi or artificial dyes such as methylene blue. The reaction is demonstrated by fluorescence, colour change when a dye is used or deposit of a dye (e.g. a blue ring of formazan from diphenyltetrazolium bromide in the presence of phenazine methosulphate).
Which screening test is used in any particular laboratory will depend on a number of factors such as cost, time required, temperature and humidity and availability of reagents. Two tests that are commonly used and that are generally reliable are described here.
Fluorescence screening test for G6PD deficiency
The method of fluorescent screening test for G6PD deficiency is that of Beutler and Mitchell modified as recommended by ICSH.
Principle
NADPH, generated by G6PD present in a lysate of blood cells, fluoresces under long-wave ultraviolet (UV) light. In G6PD deficiency, there is an inability to produce sufficient NADPH; this results in a lack of fluorescence.
Reagents
D-glucose-6-phosphate
10 mmol/l. Dissolve 305 mg of the disodium salt or an equivalent amount of the potassium salt in 100 ml of water.
NADP +
7.5 mmol/l. Dissolve 60 mg of NADP + , disodium salt, in 10 ml of water.
Saponin (white, suitable for haemolysis)
1%.
Tris-HCl buffer
750 mmol/l, pH 7.8. Dissolve 9.1 g of Tris(hydroxymethyl)aminomethane in 80 ml of water. Adjust the pH to 7.8 with HCl and make up the volume to 100 ml with water.
Oxidised glutathione (GSSG)
8 mmol/l. Dissolve 49 mg of GSSG in 10 ml of water.
Mix the reagents in the following proportion: 2 volumes of G6P, 1 volume of NADP + , 2 volumes of saponin, 3 volumes of buffer, 1 volume of GSSG and 1 volume of water.
The combined reagent is stable at − 20 °C for 2 or more years and for at least 2 months if kept at 4 °C. Azide may be added to prevent growth of contaminants without loss of activity. Dispense 100 μl volumes into appropriate small tubes and keep at − 20 °C ready for use.
Method
Thaw out sufficient tubes to set up test and controls. Allow reagents to reach room temperature before use.
Add 10 μl of whole blood (in EDTA, heparin, ACD [acid–citrate–dextrose] or CPD [citrate–phosphate– dextrose]) to 100 μl volumes of the reagent mixture and keep at room temperature (15–25 °C).
Place 10 μl of the reaction mixture on a Whatman No. 1 filter paper at the beginning of the reaction and again after 5–10 min. A shorter interval may be appropriate at a high ambient temperature ( c . 25–30 °C). Allow to air dry thoroughly before examining the spots under UV light. Record whether fluorescence is present (+) or absent (−). Always set up samples of normal blood and known G6PD-deficient blood in parallel.
If the samples are to be collected away from the laboratory or where delay is envisaged (e.g. during population screening) place about 10 μl of blood on Whatman No. 1 filter paper and allow it to dry. Cut out the disc of dried blood in the laboratory and add it to the reaction mixture. A sample of normal blood should be tested as a positive fluorescence (i.e. normal) control.
The test can be carried out on blood stored in ACD (provided it is sterile) for up to 21 days at 4 °C and for about 5 days at room temperature.
Interpretation
Fluorescence is produced by NADPH formed from NADP + in the presence of G6PD. Some of the NADPH produced is oxidised by GSSG, but this reaction, catalysed by glutathione reductase, is normally slower than the rate of NADPH production. Red cells with < 20% of normal G6PD activity do not cause detectable fluorescence.
Like all screening tests, this method is useful when large numbers of samples are to be tested, but the result must be interpreted with caution in an individual patient. The main causes of erroneous interpretations are as follows:
False-normal . If there is reticulocytosis, a vivid fluorescence may be seen with a genetically G6PD-deficient blood sample because young red cells have more G6PD activity. If the test is carried out during an acute haemolytic episode, the patient’s blood should be retested when the reticulocyte count has returned to normal.
False-deficient . If the patient is anaemic, very little fluorescence may be seen despite the G6PD being genetically normal, simply because there are relatively few red cells in the 10 μl of blood used.
Although it is possible to correct for either or both of these contingencies, if in doubt, it is best to proceed directly to a quantitative enzyme assay (discussed later).
The test is meant to give only a + or − (normal or deficient) result by comparison with the controls, and it does not make sense to grade by eye the intensity of fluorescence. If a control G6PD-deficient sample is not available, the appearance of the ‘zero time’ spot can be used for reference. The threshold for a ‘deficient’ result can be worked out by making dilutions of a normal blood sample in saline and is best set by regarding as deficient the fluorescence obtained when G6PD activity is 20% of normal or less (corresponding to a 1 in 5 dilution of normal blood). This means that very mildly deficient variants and a substantial proportion of heterozygotes (see below), will be missed. However, clinically important haemolysis is unlikely to occur in subjects who have more than 20% G6PD activity and therefore this seems an appropriate (although arbitrary) threshold for a diagnostic laboratory. Because the test depends on visual inspection, it is best to select the time of incubation in relation to ambient temperature in preliminary trials. NADPH production is a cumulative process. Therefore, given enough time, a G6PD-deficient sample will fluoresce. The time allowed for the reaction should be one at which the contrast in fluorescence between a G6PD-normal and a G6PD-deficient sample is maximal.
Methaemoglobin reduction test
Principle
Sodium nitrite converts haemoglobin to Hi. When no methylene blue is added, methaemoglobin persists, but incubation of the samples with methylene blue allows stimulation of the pentose phosphate pathway in subjects with normal G6PD levels. The Hi is reduced during the incubation period. In G6PD-deficient subjects, the block in the pentose phosphate pathway prevents this reduction.
Reagents
Sodium nitrite
180 mmol/l.
Dextrose
280 mmol/l. Dissolve 5 g of dextrose (analytical grade) and 1.25 g of NaNO 2 in 100 ml of water.
Methylene blue
0.4 mmol/l. Dissolve 150 mg of methylthionine chloride (methylene blue chloride, Sigma) in 1 litre of water.
Nile blue sulphate
22 mg in 100 ml of water. This may be used as an alternative to methylene blue.
The reagents may be used in a variety of ways to suit the convenience of the laboratory. A batch of tubes may be prepared in advance of use by mixing equal volumes of the reagents (sodium nitrite with methylene blue or Nile blue sulphate) and pipetting 0.2 ml of the combined reagent into individual glass tubes. Glass tubes must be used because plastic may adsorb some reagents. The contents of the tubes are allowed to evaporate to dryness at room temperature (15–25 °C) or in an oven at a temperature not exceeding 37 °C. The tubes must then be tightly stoppered. The reagent will keep for 6 months at room temperature. The reagents may however be used fresh, without drying.
Method
Use anticoagulated blood (EDTA or ACD) and test the samples preferably within 1 h of collection if left on the bench or within 6 h if kept at 4 °C. Blood in ACD can be stored for up to 1 week but will be unsatisfactory if there is any haemolysis. With blood from patients who are severely anaemic, adjust the PCV to 0.40 ± 0.05.
Add 2 ml of blood to the tube containing 0.2 ml of the combined reagent either freshly prepared or dried. Close the tube with a stopper and gently mix the contents by inverting it 15 times.
Prepare control tubes by adding 2 ml of blood to a similar tube without reagents (normal reference tube) and to a tube containing 0.1 ml of sodium nitrite– dextrose mixture without methylene blue (‘deficient’ reference tube).
Incubate the samples at 37 °C for 90 min. If the blood has been heparinised, incubation should be continued for 3 h.
After the incubation, pipette 0.1 ml volumes from the test sample, the normal reference tube and the deficient reference tube into 10 ml of water in separate, clear glass test tubes of identical diameter. Mix the contents gently. Compare the colours in the different tubes (see below).
Interpretation
Normal blood yields a colour similar to that in the normal reference tube (i.e. a clear red). Blood from deficient subjects gives a brown colour similar to that in the deficient reference tube. Heterozygotes give intermediate reactions.
Although this method takes longer than the fluorescent test, its advantages include the fact that it is extremely inexpensive and that the only equipment required is a water bath. In addition, the test can be complemented by cytochemical analysis that lends itself to detecting G6PD deficiency in patients with reticulocytosis and in heterozygotes.
Detection of heterozygotes for G6PD deficiency
Females heterozygous for G6PD deficiency have two populations of cells, one with normal G6PD activity and the other with deficient G6PD activity. This is the result of inactivation of one of the two X chromosomes in individual cells early in the development of the embryo. All progeny cells (i.e. somatic cells) in females will have the characteristics of only the active X chromosome. The total G6PD activity of blood in the female will depend on the proportion of normal to deficient cells. In most cases, the activity will be between 20% and 80% of normal. However, a few heterozygotes (about 1%) may have very largely normal or very largely G6PD-deficient cells.
Screening tests for G6PD deficiency fail to demonstrate most heterozygotes. The deficient red cells may, however, be identified in blood films by a cytochemical elution procedure (see below).
Test kits
Several commercial kits are available for detection of G6PD deficiency. A fluorescent spot test (Trinity Biotech 203-A) and a test based on reduction of the dye dichloroindophenol to a colourless state in the presence of phenazine methosulphate (Trinity Biotech 400) are available commercially ( www.trinitybiotech.com ).
The Quantase kit ( www.bio-rad.com ) is a photometric method for use on whole blood or dried blood spots; NADPH produced by oxidation of G6P to 6PG is measured by an increase in absorption at 340 nm.
Each test or batch of tests should include a normal and a G6PD-deficient sample. Sheep blood is a useful source of naturally deficient blood. Where possible, participation in an external quality assessment (or proficiency testing) scheme is also recommended.
Cytochemical tests for demonstrating defects of red cell metabolism
Cytochemical methods have been developed by means of which some of these defects are demonstrable in individual cells. Thus tests have been described for demonstrating red cells deficient in G6PD. The principle on which the methods are based is that red cells are treated with sodium nitrite to convert their oxyhaemoglobin (HbO 2 ) to methaemoglobin (Hi). In the presence of G6PD, Hi reconverts to HbO 2 , but in G6PD deficiency, Hi persists. The blood is then incubated with a soluble tetrazolium compound (MTT), which will be reduced by HbO 2 (but not by Hi) to an insoluble formazan form.
Attempts have been made to improve the reliability of the test for detecting heterozygotes (e.g. by controlled slight fixation of the red cells and accelerating the reaction with an exogenous electron carrier, 1-methoxyphenazine methosulphate). These cytochemical procedures are not more sensitive in the demonstration of G6PD deficiency than are the simple screening tests described above. They may, however, be useful in genetic studies and when assessing G6PD activity in women; they may be the only straightforward way to detect deficiency in the heterozygous state.
Demonstration of G6PD-deficient cells
Reagents
Sodium nitrite
0.18 mol/l (12.5 g/l). The solution must be stored in a dark bottle and made up monthly.
Incubation medium
9 g/l NaCl, 4 ml; 50 g/l glucose, 1.0 ml; 0.3 mol/l phosphate buffer, pH 7.0, 2.0 ml; 0.11 g/l Nile blue sulphate, 1.0 ml; water, 2.0 ml.
MTT tetrazolium
5 g/l of 3-(4,5-dimethyl-thiazolyl-1–2)-2,5 diphenyltetrazolium bromide in 9 g/l NaCl.
Hypotonic saline
6 g/l NaCl.
Method
Venous blood collected into ACD should be used. The test should be carried out within 8 h of collection and the blood should be kept at 4 °C until it is tested. Centrifuge the blood at 4 °C for 20 min at 1200–1500 g .
Discard the supernatant and add 0.5 ml of the packed red cells to 9 ml of 9 g/l NaCl and 0.5 ml of sodium nitrite solution contained in a 15 ml glass centrifuge tube. Incubate at 37 °C for 20 min. Centrifuge at 4 °C for 15 min at approximately 500 g , then discard the supernatant fluid without disturbing the buffy coat and uppermost layer of red cells. Wash the cells three times in cold saline. After the last washing, remove the buffy coat, mix the packed cells well and transfer 50 μl to a glass tube containing 1 ml of the incubation medium. Incubate the suspension undisturbed at 37 °C for 30 min. Then add 0.2 ml of MTT solution, shake gently and incubate at 37 °C for 1 h. Resuspend the cells thoroughly. Place one drop adjacent to one drop of hypotonic saline on a glass slide, mix the drops thoroughly and cover with a coverslip.
Examine the red cells with an oil-immersion objective, noting the presence of formazan granules ( Fig. 12-5 ).
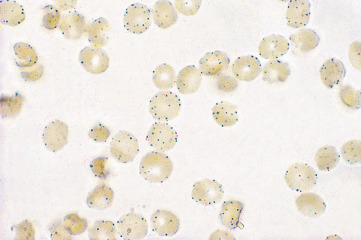
Interpretation
When G6PD activity is normal, all the red cells are stained. In hemizygotes for G6PD deficiency, the majority of the red cells are unstained. In heterozygotes, mosaicism is usually seen; usually 40–60% of the cells are unstained, but the proportion may be much less and in extreme cases as few as 2–3% may be unstained.
Pyrimidine-5′-nucleotidase screening test
Pyrimidine-5′-nucleotidase (P5N) was first described by Valentine et al . as a cytosolic enzyme in human red cells. Deficiency of P5N-1 (uridine monophosphate hydrolase-1), which shows autosomal recessive inheritance, is associated with congenital haemolytic anaemia. Heterozygotes are clinically and haematologically normal and typically have about half the normal red cell P5N activity. Homozygous P5N deficiency, in which enzyme activity is generally 5–15% of normal, results in a chronic nonspherocytic haemolytic anaemia. This is characterised by mild to moderate haemolysis, pronounced basophilic stippling visible in up to 5% of red cells and marked increase in both red cell glutathione and pyrimidine nucleotides. Osmotic fragility is normal. The rate of autohaemolysis is increased with little or no reduction in lysis by added glucose.
P5N deficiency appears to be a comparatively rare cause of hereditary nonspherocytic haemolytic anaemia. Because lead is an inhibitor of P5N, an acquired deficiency occurs in lead toxicity, and this may be important in the pathogenesis of the associated anaemia. The definitive diagnostic test is a quantitative assay of P5N activity; but the finding of supranormal levels of red cell nucleotides (mostly pyrimidines) is strongly suggestive and can be used for screening.
Activity of P5N may be measured by a colorimetric method or by a radiometric method. For the screening of P5N deficiency, the method recommended by ICSH is the determination of the UV spectra of a blood extract.
Principle
The nucleotide pool of normal red cells consists largely (> 96%) of purine (adenine and small amounts of guanine) derivatives. The levels of cytidine and uridine are normally extremely low. However, in P5N-deficient cells, more than 50% of this pool consists of pyrimidine nucleotides.
In acidic solutions, cytidine nucleotides have an absorbance maximum at approximately 280 nm, whereas adenine, guanine and uridine nucleotides absorb maximally at 260 nm. The ratio of absorbance at 260 nm to absorbance at 280 nm reflects the relative abundance of cytidine nucleotides; the absorbance ratio is lower when pyrimidine derivatives are increased.
Reagents
Sodium chloride solution
NaCl, 9 g/l.
Perchloric acid
4%. 28.6 ml of a 70% perchloric acid (PCA) solution are diluted to a final volume of 500 ml with water.
Glycine buffer
1 mol/1, pH 3.0. 7.51 g of glycine are dissolved in about 80 ml of water, the pH is adjusted to 3.0 with concentrated hydrochloric acid (HCl) and the solution is made up to a final volume of 100 ml with water.
Method
For sample preparation, centrifuge blood freshly collected in EDTA at 1200 g for 5 min, remove the plasma and wash the cells three times with ice-cold 9 g/l NaCl solution. Add 1 ml of a 50% suspension of the washed red cells to 4 ml of ice-cold 4% PCA solution and then shake vigorously for 30 s. Transfer the clear supernatant obtained after centrifugation at 1200 g for 15 min to a small test tube. Prepare a sham extract by adding 1 ml of 9 g/l NaCl to 4 ml of 4% PCA solution.
Add 500 μl of water and 300 μl of 1 mol/l glycine buffer to each of two cuvettes. To correct for optical differences between the cuvettes, read the sample cuvette against the blank at 260 and at 280 nm, giving readings B 260 and B 280 . Add 200 μl of the red cell extract to the sample cuvette and 200 μl of the sham extract to the blank cuvette. With the spectrometer zeroed at 260 nm on the blank cuvette, read the sample cuvette to obtain the value S 260 . Repeat the process at 280 nm to obtain the reading S 280 .
The A 260 /A 280 absorbance ratio (R) is calculated by subtracting the cuvette blank readings (positive or negative) at 260 and 280 nm from the readings obtained on the red cell extract when blanked against the sham extract:
R = S 260 − B 260 S 280 − B 280
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


