Chapter 18 Investigation of haemostasis
Components of normal haemostasis
The Blood Vessel
Endothelial Cell Function
The luminal surface of the endothelial cell1 is covered by the glycocalyx, a proteoglycan coat. It contains heparan sulphate and other glycosaminoglycans, which are capable of activating antithrombin, an important inhibitor of coagulation enzymes. Tissue factor pathway inhibitor (TFPI) is present on endothelial cell surfaces bound to these heparans but also tethered to a glycophosphoinositol (GPI) anchor. The relative importance of these two TFPI pools is not known. Endothelial cells express a number of coagulation active proteins that play an important regulatory role such as thrombomodulin and the endothelial protein C (PC) receptor. Thrombin generated at the site of injury is rapidly bound to a specific product of the endothelial cell, thrombomodulin. When bound to this protein, thrombin can activate PC (which degrades factors Va and VIIIa) and a carboxypeptidase which inhibits fibrinolysis (discussed later). Thrombin also stimulates the endothelial cell to produce tissue plasminogen activator (tPA). The endothelium can also synthesize protein S, the cofactor for PC. Finally, endothelium produces von Willebrand factor (VWF), which is essential for platelet adhesion to the subendothelium and stabilizes factor VIII within the circulation. VWF is both stored in specific granules called Weibel Palade bodies and secreted constitutively, partly into the circulation and partly toward the subendothelium where it binds directly to collagen and other matrix proteins. The expression of these and other important molecules such as adhesion molecules and their receptors are modulated by inflammatory cytokines. The lipid bilayer membrane also contains adenosine diphosphatase (ADPase), an enzyme that degrades adenosine diphosphate (ADP), which is a potent platelet agonist (see p. 434). Many of the surface proteins are found localized in the specialized lipid rafts and invaginations called ‘caveolae’, which may provide an important level of regulation.2
The subendothelium consists of connective tissues composed of collagen (principally types I, III and VI), elastic tissues, proteoglycans and non-collagenous glycoproteins, including fibronectin and VWF. After vessel wall damage has occurred, these components are exposed and are then responsible for platelet adherence. This appears to be mediated by VWF binding to collagen. VWF then undergoes a conformational change and platelets are captured via their surface membrane glycoprotein Ib binding to VWF. Platelet activation follows and a conformational change in glycoprotein IIbIIIa allows further, more secure, binding to VWF via this receptor as well as to fibrinogen. At low shear rates (<1000 s−1) platelet binding directly to collagen appears to dominate.3
Vasoconstriction
Vessels with muscular coats contract following injury, thus helping to arrest blood loss. Although not all coagulation reactions are enhanced by reduced flow, this probably assists in the formation of a stable fibrin plug by allowing activated factors to accumulate to critical concentrations. Vasoconstriction1,4 also occurs in the microcirculation in vessels without smooth muscle cells. Endothelial cells themselves can produce vasoconstrictors such as angiotensin II. In addition, activated platelets produce thromboxane A2 (TXA2), which is a potent vasoconstrictor.
Platelets
Platelets5,6 are small fragments of cytoplasm derived from megakaryocytes. On average, they are 1.5–3.5 μm in diameter but may be larger in some disease states. They do not contain a nucleus and are bounded by a typical lipid bilayer membrane. Beneath the outer membrane lies the marginal band of microtubules, which maintain the shape of the platelet and depolymerize when aggregation begins. The central cytoplasm is dominated by the three types of platelet granules: the δ granules, α granules and lysosomal granules. The contents of these various granules are detailed in Table 18.1. Finally there exist the dense tubular system and the canalicular membrane system; the latter communicates with the exterior. It is not clear how all these elements act together to perform such functions as contraction and secretion, which are characteristic of platelet activation.
Table 18.1 Some contents of platelet granules
| Dense (δ) granules | α Granules | Lysosomal vesicles |
|---|---|---|
| ATP | PF4 | Galactosidases |
| ADP | β-Thromboglobulin | Fucosidases |
| Calcium | Fibrinogen | Hexosaminidase |
| Serotonin | Factor V | Glucuronidase |
| Pyrophosphate | Thrombospondin | Cathepsin |
| P selectin (CD62P) | Fibronectin | Glycohydrolases |
| Transforming growth factor-beta (1) | PDGF | + others |
| Catecholamines (epinephrine/norepinephrine) | PAI-1 | |
| GDP/GTP | Histidine-rich glycoprotein | |
| α2 Macroglobulin | ||
| Plasmin inhibitor | ||
| P selectin (CD62) | ||
ADP, adenosine 5′-diphosphate; ATP, adenosine 5′-triphosphate; GDP, guanosine 5′-diphosphate; GTP, guanosine 5′-triphosphate; PAI-1, plasminogen activator inhibitor-1; PDGF, platelet-derived growth factor; PF4, platelet factor 4.
Platelet Function in the Haemostatic Process
The main steps in platelet function7 are adhesion, activation with shape change and aggregation. When the vessel wall is damaged, the subendothelial structures, including basement membrane, collagen and microfibrils, are exposed. VWF binds to collagen and microfibrils and then captures platelets via initial binding to platelet GPIb, resulting in an initial monolayer of adhering platelets. Binding via GPIb initiates activation of the platelet via a G-protein mechanism. Once activated, platelets immediately change shape from a disc to a tiny sphere with numerous projecting pseudopods. After adhesion of a single layer of platelets to the exposed subendothelium, platelets stick to one another to form aggregates. Fibrinogen, fibronectin, further VWF released from platelets and the glycoprotein Ib-IX and IIbIIIa complexes are essential at this stage to increase the cell-to-cell contact and facilitate aggregation. Certain substances (agonists) react with specific platelet membrane receptors to promote platelet aggregation and further activation. The agonists include exposed collagen fibres, ADP, thrombin, adrenaline, (epinephrine) serotonin and certain arachidonic acid metabolites including TXA2. In areas of non-linear blood flow, such as may occur at the site of an injury, locally damaged red cells release ADP, which further activates platelets.
Blood Coagulation
The central event in the coagulation pathways8 is the production of thrombin, which acts upon fibrinogen to produce fibrin and thus the fibrin clot. This clot is further strengthened by the crosslinking action of factor XIII, which itself is activated by thrombin. The two commonly used coagulation tests, the activated partial thromboplastin time (APTT) and the prothrombin time (PT), have been used historically to define two pathways of coagulation activation: the intrinsic and extrinsic paths, respectively. However, this bears only a limited relationship to the way coagulation is activated in vivo. For example, deficiencies of factor XII or of factor VIII both produce marked prolongation of the APTT, but only deficiency of the latter is associated with a haemorrhagic tendency. Moreover, there is considerable evidence that activation of factor IX (intrinsic pathway) by factor VIIa (extrinsic pathway) is crucial to establishing coagulation after an initial stimulus has been provided by factor VIIa-tissue factor (TF) activation of factor X (Fig. 18.1).8
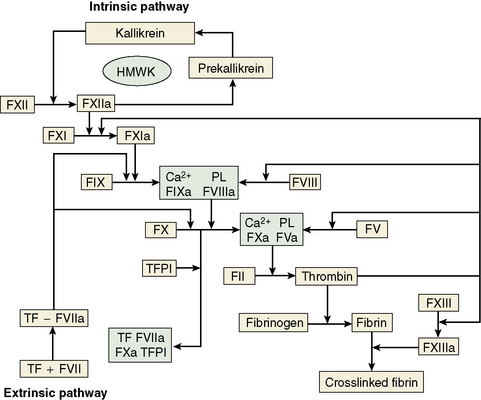
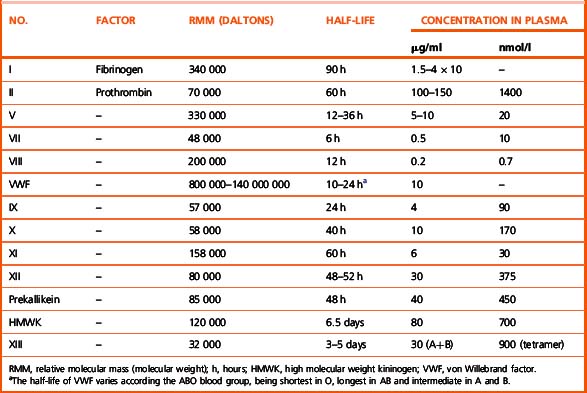
Investigation of the coagulation system centres on the coagulation factors, but the activity of these proteins is also greatly dependent on specific surface receptors and phospholipids largely presented on the surface of platelets and also by activated endothelium. The necessity for calcium in many of these reactions is frequently used to control their activity in vitro. The various factors are described in the following sections, as far as possible in their functional groups; their properties are detailed in Table 18.2.
The Contact Activation System
The contact activation system9 comprises factor XII (Hageman factor), high molecular weight kininogen (HMWK) (Fitzgerald factor) and prekallikrein/kallikrein (Fletcher factor). As mentioned earlier, these factors are not essential for haemostasis in vivo. Important activities are to activate the fibrinolytic system, to activate the complement system and to generate vasoactive peptides: in particular, bradykinin is released from HMWK by prekallikrein or FXIIa cleavage. Kallikrein and factor XIIa also function as chemoattractants for neutrophils. The contact activation system also has some inhibitory effect on thrombin activation of platelets and prevents cell binding to endothelium. Recent evidence implicates the contact system in thrombosis via activation by polyphosphate released from platelets.10
Fibrinogen
Fibrinogen11 is a large dimeric protein, each half consisting of three polypeptides named Aα, Bβ and γ held together by 12 disulphide bonds. The two monomers are joined together by a further three disulphide bonds. A variant γ chain denoted γ′ is produced by a variation in messenger RNA splicing. In the process a platelet binding site is lost and high-affinity binding sites for FXIII and thrombin are gained. The γ′ variant constitutes approximately 10% of plasma fibrinogen. A less common (<2%) γ chain variant ‘γE’ is also produced by splice variation. Fibrinogen is also found in platelets, but the bulk of this is derived from glycoprotein IIbIIIa-mediated endocytosis of plasma fibrinogen, which is then stored in alpha granules, rather than synthesis by megakaryocytes. Fibrin is formed from fibrinogen by thrombin cleavage releasing the A and B peptides from fibrinogen. This results in fibrin monomers that then associate and precipitate forming a polymer that is the visible clot. The central E domain exposed by thrombin cleavage binds with a complementary region on the outer or D domain of another monomer. The monomers thus assemble into a staggered overlapping two-stranded fibril. More complex interactions subsequently lead to branched and thickened fibre formation, making a complex mesh that binds and stabilizes the primary platelet plug.
Inhibitors of Coagulation
A number of mechanisms exist to ensure that the production of the fibrin clot is limited to the site of injury and is not allowed to propagate indefinitely.12,13 First, there are a number of proteins that bind to and inactivate the enzymes of the coagulation cascade. Probably the first of these to become active is TFPI, which rapidly quenches the factor VIIa–TF complex that initiates coagulation. It does this by combining first with factor Xa, so that further propagation of coagulation is dependent on the small amount of thrombin that has been generated during initiation being sufficient to activate the intrinsic pathway.
The Fibrinolytic System
The deposition of fibrin and its removal are regulated by the fibrinolytic system.14 Although this is a complex multicomponent system with many activators and inhibitors, it centres around the fibrinogen- and fibrin-cleaving enzyme plasmin. Plasmin circulates in its inactive precursor form, plasminogen, which is activated by proteolytic cleavage. The principal plasminogen activator (PA) in humans is tissue plasminogen activator (tPA), which is another serine protease. tPA and plasminogen are both able to bind to fibrin via the amino acid lysine. Binding to fibrin brings tPA and plasminogen into close proximity so that the rate of plasminogen activation is markedly increased and thus plasmin is generated preferentially at its site of action and not free in plasma. The second important physiological PA in humans is called urokinase (uPA). This single chain molecule (scu-PA or pro-urokinase) is activated by plasmin or kallikrein to a two-chain derivative (tcu-PA), which is not fibrin-specific in its action. However, the extent to which this is important in vivo is not clear and the identification of cell surface receptors for uPA suggests that its primary role may be extravascular. The contact activation system also appears to generate some plasminogen activation via factor XIIa and bradykinin-stimulated release of tPA. The degradation products released by the action of plasmin on fibrin are of diagnostic use and are discussed later in this chapter. The activation of plasmin on fibrin is restricted by the action of a carboxypeptidase, which removes the amino terminal lysine residues to which plasminogen and tPA bind. This carboxypeptidase is activated by thrombomodulin-bound thrombin and is referred to as thrombin-activated fibrinolysis inhibitor (TAFI).
General approach to investigation of haemostasis
This section begins with some general points regarding the clinical and laboratory approach to the investigation of haemostasis. Following this, the basic or first-line screening tests of haemostasis are described. These tests are generally used as the first step in investigation of an acutely bleeding patient, a person with a suspected bleeding tendency or as a precaution before an invasive procedure is carried out. They have the virtue that they are easily performed and the patterns of abnormalities obtained point clearly to the appropriate next set of investigations. It should be remembered, however, that these tests examine only a portion of the haemostatic mechanism and have limited sensitivity for the presence of significant bleeding diatheses such as von Willebrand disease (VWD) or disorders of platelets or vessels. Hence a normal ‘clotting screen’ should not be taken to mean that haemostasis is normal.15
Clinical Approach
The investigation of a suspected bleeding tendency may begin from three different points:
Principles of Laboratory Analysis
Assays using chromogenic peptide substrates (amidolytic assays)
The serine proteases of the coagulation cascade have narrow substrate specificities.17 It is possible to synthesize a short peptide specific for each enzyme that has a dye (p-nitroaniline, p-NA) attached to the terminal amino acid. When the synthetic peptide reacts with the specific enzyme, the dye is released and the rate of its release or the total amount released can be measured photometrically. This gives a measure of the enzyme activity present. Chromogenic substrate assays can be classified into direct and indirect assays. Direct assays can be further subclassified into primary assays, in which a substrate specific for the enzyme to be measured is used, and secondary assays, in which the enzyme or proenzyme measured is used to activate a second protease for which a specific substrate is available. Specific substrates are available for many coagulation enzymes. However, the substrate specificity is not absolute and most kits include inhibitors of other enzymes capable of cleaving the substrate to improve specificity. Indirect assays are used to measure naturally occurring inhibitors and some platelet factors.15
Coagulation assays
Coagulation assays are functional bioassays and rely on comparison with a control or standard preparation with a known level of activity. In the one-stage system optimal amounts of all the clotting factors are present except the one to be determined, which should be as near to nil as possible. The best one-stage system is provided by a substrate plasma obtained either from a patient with severe congenital deficiency or artificially depleted by immuno-adsorption. The principles of bioassay, its standardization and its limitations are considered in detail on p. 417.
Other Assays
Other assays include measurement of coagulation factors using snake venoms, assay of ristocetin cofactor and the clot solubility test for factor XIII. DNA analysis is becoming more useful and more prevalent in coagulation. However, this requires entirely different equipment and techniques (see Chapter 8).
Notes on equipment
Automated Coagulation Analysers
Pre-analytical variables including sample collection
Collection of Venous Blood
Whenever possible, venous samples should be collected without a pressure cuff, allowing the blood to enter the syringe by continuous free flow or by the negative pressure from an evacuated tube (see p. 3). Venous occlusion causes haemoconcentration, increase of fibrinolytic activity, platelet release and activation of some clotting factors. In the majority of patients, however, light pressure using a tourniquet is required; this should be applied for the shortest possible time (e.g. <1 min). The venepuncture must be ‘clean’; blood samples from an indwelling line or catheter should not be used for tests of haemostasis because they are prone to dilution and heparin contamination.
The blood is thoroughly mixed with the anticoagulant by inverting the container several times. The samples should be brought to the laboratory as soon as possible. If urgent fibrinolysis tests are contemplated, the blood samples should be kept on crushed ice until delivered to the laboratory. Assays of tPA and of PA1-1 antigen are preferably performed on samples taken into trisodium citrate to prevent continued tPA–PA1-1 binding (see p. 621).
Blood Sample Anticoagulation
The most commonly used anticoagulant for coagulation samples is trisodium citrate. A 32 g/1 (0.109 M) solution (see p. 621) is recommended. Other anticoagulants, including oxalate, heparin and ethylenediaminetetra-acetic acid (EDTA), are unacceptable. The labile factors (factors V and VIII) are unstable in oxalate, whereas heparin and EDTA directly inhibit the coagulation process and interfere with endpoint determinations. Additional benefits of trisodium citrate are that the calcium ion is neutralized more rapidly in citrate and APTT tests are more sensitive to the presence of heparin.
For routine blood coagulation testing, 9 volumes of blood are added to 1 volume of anticoagulant (i.e. 0.5 ml of anticoagulant for a 5 ml specimen). When the haematocrit is abnormal with either severe anaemia or polycythaemia, the blood:citrate ratio should be adjusted.18 For a 5 ml specimen (total), the amount of citrate should be as follows:
| Haematocrit | Citrate (ml) |
|---|---|
| 0.20 | 0.70 |
| 0.25 | 0.65 |
| 0.30 | 0.61 |
| 0.55 | 0.39 |
| 0.60 | 0.36 |
| 0.65 | 0.30 |
| 0.70 | 0.26 |
Time of Sample Collection
Desmopressin: at 30 min after intravenous infusion, 60 min after intranasal.
Calibration and Quality Control
Reference Standard (Calibrator)
International (WHO) and national standards are available for a number of coagulation factors (see p. 589). For diagnostic tests it is necessary to test a calibrated normal reference preparation alongside the patients’ plasmas.
Control Plasma
Controls are included alongside patient samples in a batch of tests. Inclusion of both normal and abnormal controls will enable detection of non-linearity in the standard curve. Whereas a reference standard (calibrator) is used for accuracy, controls are used for precision. Precision control, the recording of the day-to-day variation in control values, is an important procedure in laboratory coagulation. Participation in an external assessment scheme (see p. 594) is also important to ensure inter-laboratory harmonization. The use of lyophilized reference standard and control plasmas has become widespread, whereas locally calibrated standard pools are used especially in under-resourced countries. The results of participation in external qualitycontrol schemes require careful attention. The large number of different reagents, substrate plasmas, reference preparations and analysers available makes comparison of like with like difficult. Ideally all combinations should give similar results, but this is often not the case and the results should be used to carefully choose the combination used.
Performance of Coagulation Tests
Assay Monitoring and Endpoint Detection
Photo-Optical Analysis
 OD/min). Various wavelengths can be used such as 405 nm, 575 nm and 800 nm.
OD/min). Various wavelengths can be used such as 405 nm, 575 nm and 800 nm. OD/min). Some analysers detect light transmittance at multiple wavelengths between 395 and 710 nm.
OD/min). Some analysers detect light transmittance at multiple wavelengths between 395 and 710 nm.Percentage Detection Method
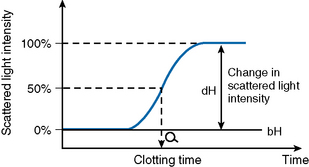
After initiating the clotting reaction, the transmitted light is monitored and a baseline A/D value (bH) is determined for the reaction (bH = 0%) (Fig. 18.2). The reaction is then monitored until the clotting reaction is completed (dH = 100%). The time to an optionally set endpoint, usually 50%, is then determined. At this point the A/D value per unit time shows the greatest change and the fibrin monomer polymerization reaction rate is high. Detection based on this principle enables coagulation analysis to be more accurate at low fibrinogen concentrations in samples with low A/D values and those samples for which the initial amount of A/D value is higher than usual, such as lipaemic and haemolysed samples.
Rate Method
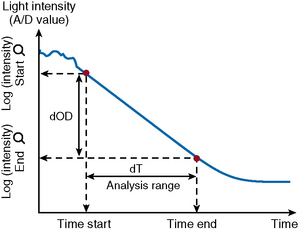
After the start of the reaction, the increase in absorbance per minute is monitored. At the predetermined end time, the final increase in absorbance per minute measurement is made. The rate of the absorbance increase per minute between these two time point measurements is calculated (Fig. 18.3). The calculated change in absorbance (dOD/min) is expressed as the raw data and used to construct a standard curve (i.e. there is no endpoint per se).
VLin Integral Method
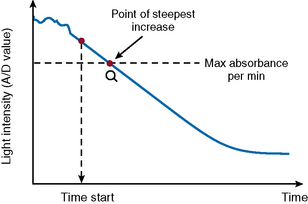
The VLin integral method (Fig. 18.4) evaluates the absorbance per minute of an immunological reaction. This is monitored and mathematical analysis used to determine the peak rate of reaction (maximum velocity). Using this method allows for increase in analytical sensitivity, extended measuring range, reduced measurement time and improved antigen excess reliability when measuring an immunological reaction. The VLin integral evaluation method is used for immunological assays, including D-dimer and VWF antigen.
Analysis Time Over
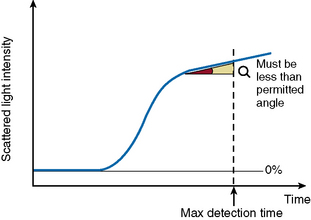
The ‘Analysis Time Over’ check (Fig. 18.5) is used to detect whether the reaction endpoint is correct. If the sample reaction end angle is greater than the permitted angle at maximum detection time, the result will be flagged with an ‘Analysis Time Over’ error. The situation occurs when testing samples with prolonged clotting times and satisfactory endpoint has not been reached by the end of the time allotted for analysis. When this occurs the following checks should be performed:
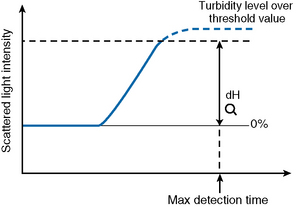
Turbidity Level Over
If the dH exceeds the detection capacity of the A/D converter, the result will not be reported and it may be suspected that sample plasma is turbid or lipaemic (Fig. 18.6). When this occurs, the following checks should be performed:
Clot Signatures: Normal and Abnormal APTT Clot Waveforms
Information on the dynamics of clot formation may also be extracted from the optical profiles generated when performing the PT or APTT tests. It has been demonstrated that such profiles (clot waveforms) show a different pattern in certain clinical conditions compared to normal (Fig. 18.7). Furthermore, the shape of this pattern is predictable for the particular abnormality and the term ‘clot signature’ has been used in this context.
The A2 Flag on the MDA system identifies the presence of a biphasic APTT waveform often seen in patients with DIC and a high sensitivity (98%), specificity (98%) and positive predictive value (74%) have been reported.19