Chapter 14 Investigation of abnormal haemoglobins and thalassaemia
The haemoglobin molecule
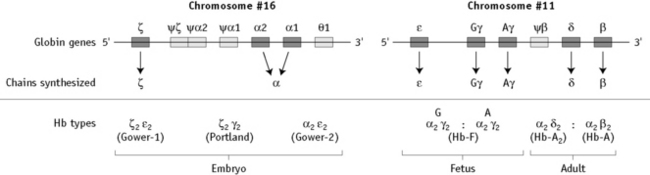
α chain synthesis is directed by two α genes, α1 and α2, on chromosome 16 and β and δ chain synthesis by single β and δ genes on chromosome 11; γ chain synthesis is directed by two genes, Gγ and Aγ, also on chromosome 11. The globin genes are shown diagrammatically in Figure 14.1.
There are many naturally occurring, genetically determined variants of human haemoglobin (>1000)1 and although many are harmless, some have serious clinical effects. Collectively, the clinical syndromes resulting from disorders of haemoglobin synthesis are referred to as ‘haemoglobinopathies’. They can be grouped into three main categories:
An individual can also have a combination of two or more of these abnormalities.
Structural variants of haemoglobin
Haemoglobins with Reduced Solubility
Hb S
By far the most common haemoglobin variant in this group is sickle haemoglobin or Hb S. As a result of the replacement of glutamic acid by valine in position 6 of the β chain, Hb S has poor solubility in the deoxygenated state and can polymerize within the red cells. The red cell shows a characteristic shape change because of polymer formation and becomes distorted and rigid, the so-called sickle cell (see p. 90, Fig. 5.71). In addition, intracellular polymers lead to red cell membrane changes, generation of oxidant substances and abnormal adherence of red cells to vascular endothelium.
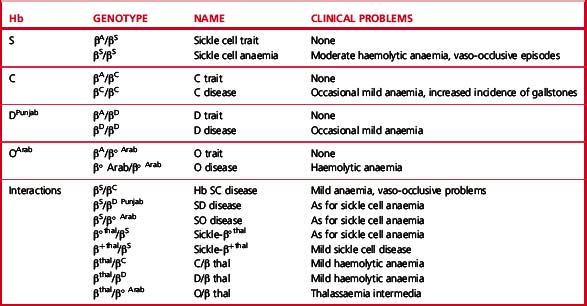
Clinical syndromes associated with common structural variants and those owing to their interaction with β thalassaemia are shown in Table 14.1.
Sickle Cell Disease
Sickle cell disease2 (also called disorder) is a collective name for a group of conditions causing clinical symptoms that result from the formation of sickle red cells. It is common in people originating from Africa, but it is also found in considerable numbers of people of Indian, Arabic and Greek descent.
The homozygous state or sickle cell anaemia (β genotype SS) causes moderate anaemia resulting both from haemolysis and from the reduced oxygen affinity of haemoglobin S. The main clinical disability arises from repeated episodes of vascular occlusion by sickled red cells resulting in acute crises and eventually in end-organ damage. The clinical severity of sickle cell anaemia is extremely variable. This is partly due to the effects of inherited modifying factors, such as interaction with α thalassaemia or increased synthesis of Hb F and partly to socioeconomic conditions and other factors that influence general health.2
Other Forms of Sickle Cell Disease
Sickle β/thalassaemia arises as a result of inheritance of one Hb S and one β thalassaemia gene. Africans and Afro-Caribbeans with this condition are often heterozygous for a mild β+ thalassaemia allele resulting in the production of about 20% of Hb A. This gives rise to a mild sickling disorder. Inheritance of Hb S and β° thalassaemia trait is associated with severe sickle cell disease. Interaction of Hb S with haemoglobin DPunjab (Hb DLos Angeles) or with Hb OArab gives rise to severe sickle cell disease.2
Hb C
Hb C is the second most common structural haemoglobin variant in people of African descent. The substitution of glutamic acid in position 6 of the β chain by lysine results in a haemoglobin molecule with a highly positive charge, decreased solubility and a tendency to crystallize. However, Hb C does not give a positive sickle solubility test. Heterozygotes are asymptomatic, but target cells and irregularly contracted cells may be present in blood films. Homozygotes may have mild anaemia with numerous target cells and irregularly contracted cells (see Chapter 5, Fig. 5.32). Interaction with β° or β+ thalassaemia trait results in mild or moderate haemolytic anaemia.
Unstable Haemoglobins
Amino acid substitutions close to the haem group, or at the points of contact between globin chains, can affect protein stability and result in intracellular precipitation of globin chains. The precipitated globin chains attach to the red cell membrane giving rise to Heinz bodies, and the associated clinical syndromes were originally called the congenital Heinz body haemolytic anaemias. Changes in membrane properties may lead to haemolysis, often aggravated by oxidant drugs. There is considerable heterogeneity in the haematological and clinical effects of unstable haemoglobins. Many are almost silent and are detected only by specific tests, whereas others are severe, causing haemolytic anaemia in the heterozygous state. Hb Köln is the most common variant in this rare group of disorders.3,4
Haemoglobins with Altered Oxygen Affinity
Haemoglobin variants with altered oxygen affinity are a rare group of variants that result in increased or reduced oxygen affinity.5 Mutations that increase oxygen affinity are generally associated with benign lifelong erythrocytosis. This may be confused with polycythaemia vera and may be inappropriately treated with cytotoxic drugs or 32P.
Hb M
The Hb M group is another rare group of variants.6 Such haemoglobins have a propensity to form methaemoglobin, generated by the oxidation of ferrous iron in haem to ferric iron, which is incapable of binding oxygen. Despite marked cyanosis, there are few clinical problems. Most are associated with substitutions that disrupt the normal six-ligand state of haem iron.
Thalassaemia syndromes
The thalassaemia syndromes7 are a heterogeneous group of inherited conditions characterized by defects in the synthesis of one or more of the globin chains that form the haemoglobin tetramer. The clinical syndromes associated with thalassaemia arise from the combined consequences of inadequate haemoglobin production and of unbalanced accumulation of one type of globin chain. The former causes anaemia with hypochromia and microcytosis; the latter leads to ineffective erythropoiesis and haemolysis. Clinical manifestations range from completely asymptomatic microcytosis to profound anaemia that is incompatible with life and can cause death in utero (Table 14.2). This clinical heterogeneity arises as a result of the variable severity of the primary genetic defect in haemoglobin synthesis and the coinheritance of modulating factors, such as the capacity to synthesize increased amounts of Hb F.
Table 14.2 Clinical syndromes of thalassaemia
| Clinically asymptomatic |
| Silent carriers |
| α+ thalassaemia heterozygotes (some cases) |
| Rare forms of β thalassaemia trait |
| Thalassaemia minor (low MCH and MCV, with or without mild anaemia) |
| α+ thalassaemia heterozygotes (some cases) |
| α° thalassaemia heterozygotes |
| α+ thalassaemia homozygotes |
| β° thalassaemia trait |
| β+ thalassaemia trait |
| Some cases of Hb E/β thalassaemia |
| Thalassaemia intermedia (transfusion independent)a |
| Some β+/β+ thalassaemia homozygotes and compound heterozygotes |
| Interaction of β°/β°, β°/β+ or β+/β+ with |
| α thalassaemia |
| Interaction of β°/β or β+/β with triple α |
| Hb H disease |
| α°/Hb Constant Spring thalassaemia |
| β°/δβ or β+/δβ thalassaemia compound heterozygotes |
| δβ/δβ thalassaemia |
| Some cases of Hb E/β thalassaemia and Hb Lepore/β thalassaemia |
| Rare cases of heterozygotes for β thalassaemia mutation, particularly involving exon 3 (‘dominant β thalassaemia’) |
| Thalassaemia major (transfusion dependent) |
| β°/β° thalassaemia |
| β+/β+ thalassaemia |
| β°/β+ thalassaemia |
| Some cases of β°/Hb Lepore and β+/Hb Lepore thalassaemia |
| Some cases of β°/Hb E and β+/Hb E thalassaemia |
MCH, mean cell haemoglobin; MCV, mean cell volume.
a β thalassaemia intermedia is defined as a symptomatic condition in which regular transfusion is not essential to maintain life; however some patients have a poor quality of life and may benefit from transfusion as their condition progresses.
Thalassaemias are generally inherited as alleles of one or more of the globin genes located on either chromosome 11 (for β, γ and δ chains) or on chromosome 16 (for α chains). They are encountered in every population in the world but are most common in the Mediterranean littoral and near equatorial regions of Africa and Asia. Gene frequencies for the α and β thalassaemias on a global basis range from 1% to more than 80% in areas where malaria is endemic.8
β Thalassaemia Syndromes
Many different mutations cause β thalassaemia and related disorders.9 These mutations can affect every step in the pathway of globin gene expression: transcription, processing of the messenger ribonucleic acid (mRNA) precursor, translation of mature mRNA and preservation of post-translational integrity of the β chain. More than 200 mutations have been described.10 Most types of β thalassaemia are the result of point mutations affecting the globin gene, but some large deletions are also known. Certain mutations are particularly common in some communities. This helps to simplify prenatal diagnosis, which is carried out by detection or exclusion of a particular mutation in fetal DNA.
The effect of different mutations varies greatly. At one end of the spectrum are a group of rare mutations, mainly involving exon 3 of the β globin gene, which are so severe that they can produce the clinical syndrome of thalassaemia intermedia in the heterozygous state. At the other end are mild alleles that produce thalassaemia intermedia in the homozygous or compound heterozygous state and some that are so mild that they are completely haematologically silent, with normal mean cell volume (MCV) and Hb A2 in the heterozygous state. In between are the great majority of β+ and β° alleles, which cause β thalassaemia major in the homozygous or compound heterozygous state and in the heterozygous state give rise to a mild anaemia (or Hb at the low end of the normal range), with microcytic, hypochromic indices and raised Hb A2.11
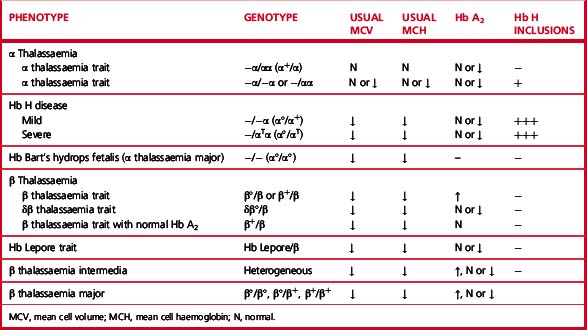
Heterozygotes for β thalassaemia alleles usually have either a normal haemoglobin with microcytosis or a mild microcytic hypochromic anaemia; Hb A2 is elevated and Hb F is sometimes also elevated. Laboratory features of various β thalassaemia syndromes are shown in Table 14.3.
α Thalassaemia Syndromes
There are four syndromes of α thalassaemia:12 α+ thalassaemia trait, where one of the two globin genes on a single chromosome fails to function; α° thalassaemia trait, where two genes on a single chromosome fail to function; Hb H disease, with three genes affected; and Hb Bart’s hydrops fetalis, where all four are absent or defective. These syndromes are usually a result of deletions of one or more genes, although approximately 20% of the mutations described are non-deletional. α+ thalassaemia is particularly common in Africa and α° thalassaemia is common in South-east Asia. The laboratory features are shown in Table 14.3.
Hb H disease gives rise to haemolytic anaemia; patients rarely require transfusion or splenectomy.

Increased Hb F in Adult Life
Haemoglobin production in man is characterized by two major switches in the haemoglobin composition of the red cells. During the first 3 months of gestation, human red cells contain embryonic haemoglobins (see p. 302), whereas during the last 6 months of gestation, red cells contain predominantly fetal haemoglobin. The major transition from fetal to adult haemoglobin synthesis occurs in the perinatal period and by the end of the first year of life red cells have a haemoglobin composition that usually remains constant throughout adult life. The major haemoglobin is then Hb A, but there are small amounts of Hb A2 and Hb F. Only 0.2–1.0% of total haemoglobin in human red cells is Hb F and it is restricted to a few cells called ‘F’ cells. Both the number of F cells and the amount of Hb F per cell can be increased in various conditions, particularly if there is rapid bone marrow regeneration.13
The general organization of human globin gene clusters is shown in Figure 14.1. The products of two γ genes differ in only one amino acid: Gγ has glycine in position 136, whereas Aγ has alanine. In fetal red cells, the ratio of Gγ to Aγ is approximately 3:1; in adult red cells, it is approximately 2:3.
Inherited Abnormalities That Increase Hb F Concentration
More than 50 mutations that increase Hb F synthesis have been described.13,14 They result in one of two phenotypes, HPFH or δβ thalassaemia; differentiation between these two types is not always simple but has clinical relevance. In general, HPFH has a higher percentage of Hb F and much more balanced chain synthesis. The most common, the African type of HPFH, is associated with a high concentration of Hb F (15–45%), pancellular distribution of Hb F on Kleihauer staining and normal red cell indices. Mutations causing increased synthesis of Hb F are mostly deletions, but some non-deletion mutations have also been described. In contrast, subjects with δβ thalassaemia have lower levels of Hb F accompanied by microcytic, hypochromic indices. The major clinical significance of these abnormalities is their interaction with β thalassaemia and Hb S. Compound heterozygotes for either of these conditions and HPFH have much milder clinical syndromes than the homozygotes for haemoglobin S or β thalassaemia. Compound heterozygotes for either of these conditions and δβ thalassaemia have a condition much closer in severity to the homozygous states.
Investigation of patients with a suspected haemoglobinopathy
Investigation of a person at risk of a haemoglobinopathy encompasses the confirmation or exclusion of the presence of a structural variant, thalassaemia trait or both. If a structural haemoglobin variant is present, it is necessary to ascertain the clinical significance of the particular variant so that the patient is appropriately managed. If it is confirmed that thalassaemia trait is present, it is not usually necessary to determine the precise mutation present because the clinical significance is usually negligible. The exception to this is an antenatal patient whose partner has also been found to have thalassaemia trait. If prenatal diagnosis is being considered, it may be necessary to undertake mutation analysis to predict fetal risk accurately and to facilitate prenatal diagnosis (see p. 146).
In the majority of patients, the presence of a haemoglobinopathy can be diagnosed with sufficient accuracy for clinical purposes from knowledge of the patient’s ethnic origin and clinical history (including family history) and the results of physical examination combined with relatively simple haematological tests. Initial investigations should include determination of haemoglobin concentration and red cell indices. A detailed examination of a well-stained blood film should be carried out. In some instances, a reticulocyte count and a search for red cell inclusions give valuable information. Assessment of iron status by estimation of serum iron and total iron binding capacity and/or serum ferritin is sometimes necessary to exclude iron deficiency. Other important basic tests are haemoglobin electrophoresis or high-performance liquid chromatography (HPLC), a sickle solubility test and measurement of Hb A2 and Hb F percentage. In cases of common haemoglobin variants and classical β thalassaemia trait, accurate data from these tests will facilitate a reliable diagnosis without the need for more sophisticated investigations. However, definitive diagnosis of some thalassaemia syndromes can only be obtained using DNA technology (see p. 147 and p. 330). Similarly, in particular situations, haemoglobin variants will require unequivocal identification by the use of DNA technology or protein analysis by mass spectrometry.15 Individuals or families who require such investigation must be carefully selected on the basis of family history and on the results of the basic investigations described later in this chapter. Large-scale screening programmes are increasingly being undertaken in some countries where individual case histories and the results of other laboratory tests are not usually available. The problems of such programmes are discussed on p. 318.
Laboratory investigation of a suspected haemoglobinopathy should follow a defined protocol, which should be devised to suit individual local requirements. The data obtained from the clinical findings, blood picture and electrophoresis or HPLC will usually indicate in which direction to proceed. The investigation for a structural variant is described in the first section and that for a suspected thalassaemia syndrome is described in the second section of this chapter. Screening tests for thalassemia trait and haemoglobin E trait that may be especially applicable in under-resourced areas are described in Chapter 26.
Laboratory detection of haemoglobin variants
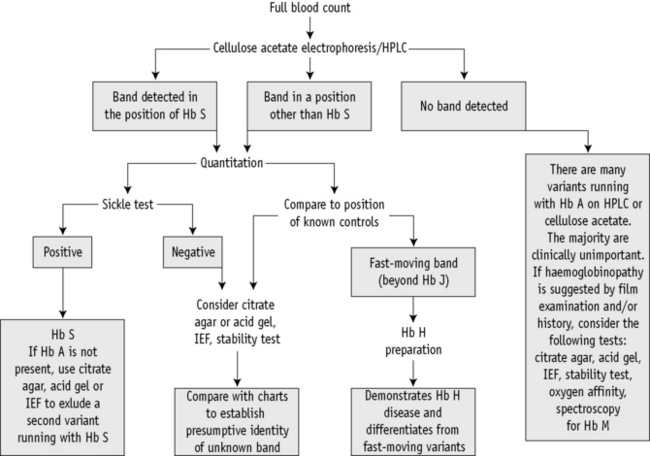
A proposed scheme of investigation is shown in Figure 14.2 and a list of procedures follows:15,16
Blood Count and Film
The blood count, including Hb and red cell indices, provides valuable information useful in the diagnosis of both α and β thalassaemia interactions with structural variants (see Chapter 3). A film examination may reveal characteristic red cell changes such as target cells in Hb C trait, sickle cells in sickle cell disease and irregularly contracted cells in the presence of an unstable haemoglobin (see Chapter 5).
Discriminant functions using various formulae have been proposed as a basis for further testing for thalassaemia,17,18 but we do not advise their use. Although such functions and formulae do indicate whether thalassaemia or iron deficiency is more likely, they may lead to individuals who have both iron deficiency and thalassaemia trait not being tested promptly. Generally this is not a problem and indeed it may be preferable to keep the patient under observation until iron deficiency has been treated and then to reassess the likelihood of thalassaemia trait. However, many of the patients who require testing for thalassaemia are women who are already pregnant. In such patients the likely delay in testing is unacceptable. Moreover, these formulae do not appear to have been validated for use during pregnancy. For these reasons, we advise that whenever genetic counselling might be required, testing for β thalassaemia trait should be carried out in all individuals with an MCH <27 pg and screening for α thalassaemia trait should be carried out in those individuals with an MCH <25 pg who belong to an ethnic group in which α° thalassaemia is prevalent.11
Collection of Blood and Preparation of Haemolysates
EDTA is the most convenient anticoagulant because it is used for the initial full blood count and film (see Chapter 1), although samples taken into any anticoagulant are satisfactory. Cells freed from clotted blood can also be used.
Control Samples
Note: Repeated freezing and thawing should be avoided.
Quality Assurance
Because the haemoglobinopathies are inherited conditions, some of which carry considerable clinical and genetic implications, precise documentation and record-keeping are of paramount importance.19 The use of cumulative records when reviewing a patient’s data is very useful because it of itself constitutes an aspect of quality assurance. In some situations, repeat sampling, family studies or both may be required to elucidate the nature of the abnormality in an individual.
In-house standard operating procedures should be followed carefully, particularly in this field of haematology, where a small difference in technique can make a significant difference in the results obtained and can lead to misdiagnosis. Many of the techniques described have attention drawn to specific technical details that are important for ensuring valid results.20
It is necessary to use reference standards and control materials in each of the analyses undertaken and in some cases to use duplicate analysis to demonstrate precision. There are international standards for Hb F and Hb A2 (see p. 591), whereas in some countries national reference preparations are also available from national standards institutions. These are extremely valuable because the target values have been established by collaborative studies. Control materials can be prepared in-house or obtained commercially. Samples stored as whole blood at 4°C can be used reliably for several weeks. All laboratories should confirm the normal range for their particular methods and the normal range obtained should not differ significantly from published data.
All laboratories undertaking haemoglobin analysis should participate in an appropriate proficiency testing programme (see p. 594). In the UK, the National External Quality Assessment Scheme (NEQAS) provides samples for sickle solubility tests; for detection and quantitation of variant haemoglobins; and for quantitation of Hbs A2, F and S.
National and international guidelines have been published for all aspects of the investigations given here.11,21–23,20,24,25
Cellulose Acetate Electrophoresis at Alkaline pH
Haemoglobin electrophoresis at pH 8.4–8.6 using cellulose acetate membrane is simple, reliable and rapid. It is satisfactory for the detection of most common clinically important haemoglobin variants.23–25
Reagents
Fixative/stain solution. Ponceau S, 5 g; trichloroacetic acid, 7.5 g; water to 1 litre
Destaining solution. 3% (v/v) acetic acid, 30 ml; water to 1 litre
Haemolysing reagent. 0.5% (v/v) Triton X-100 in 100 mg/l potassium cyanide.
Method


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


