STEREOTYPIC PATTERNS OF METASTASIS TO DISTANT ORGANS BY CANCER TYPE

Heterogeneity in Cancer Metastasis and Rarity of Metastatic Cells
Because numerous sequential steps are needed for metastasis, multiple genetic changes are envisioned. A failure in any step would prevent metastasis altogether. Accordingly, tumor cells that can accumulate a full complement of needed alterations to endow them with metastatic ability should be rare. These ideas are supported by early experiments. Work by Fidler and colleagues7 showed that subpopulations of tumor cells exist that display significant variation in their metastatic ability and metastatic lesions likely arose from single progenitor cells. Early cell fate studies revealed that less than 0.01% of tumor cells gave rise to metastases. More recent studies using in vivo video microscopy to visualize and quantitate cell fate confirmed that metastasis is an inefficient process (reviewed in ref. 8). Thus, important early studies helped to establish the idea that primary tumors are heterogeneous in their metastatic ability and that tumor cells that can successfully metastasize are exceedingly rare.
The Traditional Progression Model for Metastasis and Its Implications
A synthesis of clinical observation, deduced steps in the metastatic cascade, and early studies of experimental metastasis in mice led to a traditional model for metastatic progression.7 In this view, primary tumor cells undergo somatic evolution and accumulate genetic changes. Because numerous steps are required for metastasis, the number of genetic changes that are needed for full metastatic competency is large; hence, tumor cells that have acquired these changes are rare. Many clinicopathologic traits such as lymphovascular invasion and regional lymph node involvement represent successful completion of some of the steps in the metastatic cascade but not necessarily all. The clinical observation that metastatic risk increases with tumor size is explained by mathematical considerations predicting that genetic changes accumulate faster with increased population size. Larger tumors are more likely to contain rare cells that are metastatically competent, making metastasis a late event in tumorigenesis.
One of the primary objectives in the clinical management of cancer is to prevent or decrease the risk of metastasis. How this objective is approached is shaped by empiricism and perceptions about how metastasis proceeds. The idea that metastasis occurs as a late event in tumorigenesis argues that early detection and early eradication of the primary tumor will prevent metastasis and be sufficient for cure. Screening programs, radical versus more limited surgical excisions, and the use of adjuvant radiation to the surgical bed can be justified based on the idea that cancers caught early have not likely spread. Metastatic heterogeneity within the primary tumor and the rarity of tumor cells that can complete all the sequential steps in the metastatic cascade suggest that the detection of tumor cells caught in the act of undergoing an early step in the cascade may still represent an opportunity to stop metastasis in its tracks. This is a rationale for oncologic surgeries that include regional lymph node dissections and the use of regional radiation therapy. The likely emergence of rare metastatic cells late during tumorigenesis provides reason to add adjuvant systemic chemotherapy after local treatment of larger and more advanced primary tumors rather than smaller tumors with less aggressive features.
Alternative Models
Although the traditional model for metastasis has enjoyed favor, alternative models have been proposed. The clinical data for breast cancer has inspired a long-standing debate on whether metastasis follows a traditional progression model or a predetermination paradigm—also known as the Halsted model versus the Fisher model (discussed in ref. 9) for metastasis.9 Both models seek to justify and explain clinical data looking at the benefit of aggressive local treatment of the primary tumor and draining lymph nodes versus the early use of adjuvant systemic chemotherapy. Although more anatomic than cellular in nature, the Halsted model looked at breast cancer from a traditional vantage point and imposed on it an orderly anatomic spread pattern from primary site, to regional lymph nodes, to distant organs. This orderly progression would make complete eradication of the primary and regional tumor burden sufficient to stop metastasis. In contrast, Fisher hypothesized that whether distant relapse occurs in breast cancer is predetermined from the onset of tumorigenesis (discussed in ref. 9). This view emphasizes breast cancer as a systemic disease for those tumors so fated and the importance of adjuvant systemic chemotherapy. The data from randomized trials for adjuvant treatment and from breast cancer screening programs do not clearly rule-out one model or the other.10 To reconcile the clinical data, Hellman9 proposed that breast cancer is best considered a spectrum of diseases bound by predetermination models and traditional progression models. Other models that conceptually differ from the traditional progression model include the clonal dominance model11 and the dynamic heterogeneity model.12
Compatibility of Metastasis Models with Somatic Evolution
Both alternative and traditional progression models alike need to be compatible with the paradigm of somatic evolution, which presents a potential problem. Because it is not obvious why metastasis genes that promote growth at a distant site should have a fitness advantage for a primary tumor, the likelihood that multiple metastasis-specific genes will become fixed in a primary tumor would seem unlikely. To reconcile this, it has been suggested that the genes selected to drive primary tumor formation and progression are also the genes that mediate metastasis.13 This notion would imply that metastasis is a predetermined property of primary tumors that principally depends on the history of oncogenes and tumor suppressor genes that the primary tumor acquires. Such early onset of metastatic ability could explain phenomenon like cancers of unknown primary and support earlier predetermination metastasis models for breast cancer. However, as previously mentioned, predetermination models are not always consistent with clinical data, in particular the ability of screening and early detection to decrease cancer mortality. Furthermore, the phenomenon of metastatic dormancy, whereby metastasis remains inactive and undetectable for years if not decades after treatment of the primary tumor, is difficult to explain unless further metastasis-promoting changes occur after the primary tumor has been removed.
AN INTEGRATED MODEL FOR METASTASIS
Different concepts on how metastasis progresses have individual merits and limitations. A clearer understanding of metastasis requires sophisticated insight on a molecular level. Recent advances in the field of metastasis research are beginning to bring together an integrated and more complex paradigm (Fig. 10.1) whereby elements from different models may be interconnected.14 At the heart of this integrated paradigm are the principles of somatic evolution. Somatic evolution selects for functions and not directly for specific genes. Therefore, during primary tumor growth, the principal functions that are selected are tumorigenic functions that can be met by a large repertoire of oncogenic mutations. Examples of these tumorigenic functions include proliferative and metabolic autonomy, self-renewal ability, resistance to cell death, resistance to inhibitory signals, immune evasion, motility, invasion, and angiogenesis. Most of these traits were enumerated by Hanahan and Weinberg15 as being hallmarks of cancer. Many of these tumorigenic functions allow transformed cells to attract supporting stroma and migrate and invade surrounding tissue, regardless of whether or not cells reside in the primary tumor. This subset of tumorigenic functions is a prerequisite for metastasis because such functions are needed for cells to invade, penetrate blood vessels, and give rise to circulating tumor cells. These functions are shared by primary tumors and metastasis and are defined as metastasis initiation functions. A prominent example includes epithelial-to-mesenchymal transition (EMT).
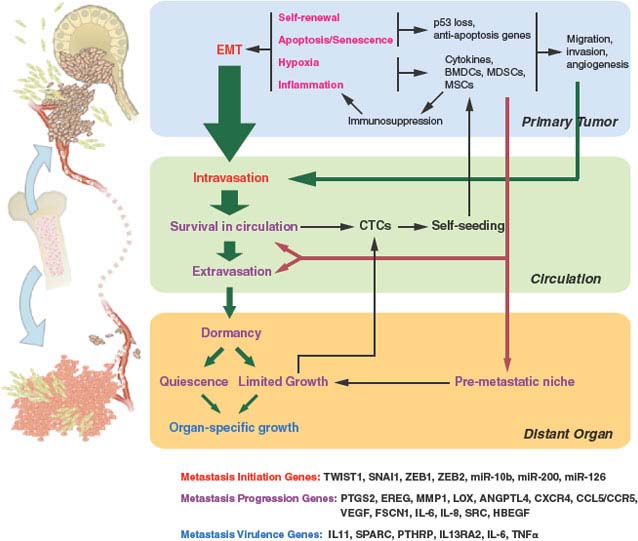
FIGURE 10.1 Selective pressures and steps from primary tumor growth to metastasis. Selective pressures at the primary tumor (labeled in magenta) can determine metastatic potential. Cancer is initiated by oncogenic changes. Of particular relevance to metastatic potential may be self-renewal pathways and the need to overcome apoptosis and senescence. Hypoxia and inflammation have important roles and lead to tumor cells co-opting bone marrow-derived cells (BMDCs), myeloid-derived suppressor cells (MDSCs), and mesenchymal stem cells (MSCs), to name a few. These cells and the cytokines that they produce enhance the ability of the tumor to migrate, invade, overcome hypoxia, and maintain an immunosuppressed environment. The ability of primary tumor cells to undergo an epithelial-to-mesenchymal transition (EMT) is also influenced by the selective pressures faced during primary tumor growth. EMT results in migration, invasion, and intravasation. Such functions (labeled in red) are examples of metastasis initiation functions. Although non−EMT-related migration and invasion can also lead to intravasation, EMT is likely a principle means by which circulating tumor cells (CTCs) are promoted. Further steps in the metastatic cascade are shown by green arrows, with the size of the arrow representing the likelihood that the step is successfully completed for many cancer types (e.g., breast cancer). Selective pressures encountered from the local microenvironment shape metastatic proclivity by selecting for tumorigenic functions that secondarily help cells navigate the metastatic cascade (metastasis-progression functions). Hypoxia and inflammation-related events contribute to metastasis-progression functions (labeled in purple) by enhancing the ability of CTCs to survive in the circulation, extravasate, and form a premetastatic niche. CTCs can either self-seed the primary tumor, which results in augmentation of tumor mass and further selection of metastatic traits, or extravasate into distant organs. Although a premetastatic niche facilitates adaptation to the foreign microenvironment of distant organs, further selection is needed for full colonization. This results in a period of dormancy whereby micrometastases remain quiescent or growth is counterbalanced by apoptosis and the lack of angiogenesis. Further somatic evolution within the distant organ can eventually result in selection of macrometastatic-colonization functions (labeled in blue) and organ-specific growth. On the bottom of the figure are examples of specific genes that play a role in metastasis initiation, metastasis progression, and macrometastatic colonization.
It is evident how genes with tumorigenic functions and genes with metastasis initiation functions can be selected for during primary tumor growth. However, how are metastasis-specific functions (i.e. functions that are not characteristic of general tumorigenesis) selected during growth at the primary site? Metastasis-specific functions include survival in the circulation, extravasation, survival in the microenvironment of distant organs, and organ-specific colonization. Recent experimental evidence reveals that some genes can mediate tumorigenic functions and secondarily serve metastasis-specific functions either in a general way or with particular organ selectivity.16,17 These types of functions are called metastasis-progression functions and genes with this duality are defined as metastasis-progression genes. Metastasis-progression genes form the basis for predetermination models for metastasis. When metastasis-progression genes are selected for, their expression by the primary tumor will track with increased risk of metastasis. These genes will also mechanistically couple certain traits of primary tumor progression (e.g., rapid growth, invasiveness, resistance to hypoxia) with distant spread.
Cancer cells that have acquired metastasis-progression genes can undergo additional selective pressure during life away from the primary tumor. Functionally, genes selected by the pressures of a distant site are similar to metastasis-progression genes but they are not coupled to tumorigenic genes and so confer no advantage to a primary tumor. Therefore, altered expression of these genes would be rare or absent in the primary tumor and discernible only in the metastatic lesion. These genes are called macrometastatic-colonization genes and provide macrometastatic-colonization functions. Macrometastatic-colonization genes form the basis of traditional progression models for metastasis.
In this integrated view that stratifies genes into tumorigenic, metastasis initiation, metastasis progression, and macrometastatic colonization, the selection for tumorigenic functions during primary tumor growth provides essential prerequisites for future metastasis. Certain biases in the genes that are selected to fulfill particular tumorigenic functions may result in genes that can also fulfill specific metastatic functions, leading to an early proclivity toward distant spread. The further selection of metastasis-specific functions after infiltration of distant organs can continue to modify metastatic behavior through the acquisition of macrometastatic-colonization genes. Although the emerging evidence does not always allow clear delineation between genes that serve tumorigenic versus metastasis initiation versus metastasis-progression functions, recent molecular understanding and insight offer the underpinnings of this integrated view.
SELECTIVE PRESSURES AT THE PRIMARY TUMOR DRIVING ACQUISITION OF METASTASIS FUNCTIONS
Of all the tumorigenic functions required by aggressive primary cancers, several may additionally select for metastasis initiation or metastasis-progression genes (Fig. 10.1). Experimental and clinical evidence point toward the following factors.
Hypoxia
In order to disrupt tissue homeostasis during primary tumorigenesis, many barriers that can limit growth must be overcome. A near-universal need is for tumors to respond to hypoxia (reviewed in refs. 18 through 20). Normal tissue such as epithelium is separated from blood vessels by a basement membrane. When preinvasive tumor growth occurs, hypoxia can ensue because oxygen and glucose typically can only diffuse 100 to 150 microns, resulting in portions of the expanding mass becoming hypoxic. This can be seen in comedo-type ductal carcinoma in situ (DCIS) of the breast, whereby a necrotic center characterizes these preinvasive breast tumors. The fact that DCIS can take years to progress to invasive cancer, or never progresses to cancer, suggests that hypoxia can be a significant barrier.
Although there are multiple paths that cancer cells can take to adapt to hypoxia, the hypoxia-inducible factor (HIF) transcription factors have a central role. Under hypoxic conditions, HIF-1α and HIF-2α become stabilized, resulting in the transcription of over 100 HIF-α regulated genes. These target genes are involved in angiogenesis, glycolysis, and invasion, which together help hypoxic cells adapt. Up-regulated angiogenesis genes include vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF). These factors cause quiescent blood vessels to undergo remodeling, including the laying down of a matrix that activated endothelial cells use to form newly vascularized areas. Various glycolysis genes are expressed and their metabolic by-products lead to acidification of the extracellular space. This is normally toxic to cells and requires further adaptation either by up-regulation of H+ transporters or acquired resistance to apoptosis. To assist in invasion toward newly vascularized areas, HIF-α up-regulates matrix metalloproteinase 1 and 2 (MMP1, MMP2), lysyl oxidase (LOX), and the chemokine receptor CXCR4. Degradation of the basement membrane by MMP2 and alteration of the extracellular matrix (ECM) by MMP1 and LOX clears away a barrier to migration. The activation of CXCR4 then stimulates cancer cells to migrate to regions of angiogenesis. Thus, if these series of events can be successfully completed, not only will preinvasive tumors successfully deal with hypoxia, but they will also likely invade through the basement membrane in the process. Invasion through the basement membrane defines invasive carcinomas.
Inflammation
When normal tissue homeostasis and architecture are disrupted, this can lead to vessel injury, hypoxic zones, extravasation of blood proteins, and the entry of foreign pathogens (reviewed in refs. 21 and 22). A rapid response is mounted by a front line composed of immune and bone marrow-derived cells (BMDCs) such as lymphocytes, neutrophils, macrophages, dendritic cells, eosinophils, and natural killer (NK) cells. The purpose is to restore homeostasis through several phases: inflammation, tissue formation, and tissue remodeling. In the initial phase, tissue breakdown attracts neutrophils to infiltrate the wounded area and release various proinflammatory cytokines such as interleukin (IL)-8), IL-1β, and tumor necrosis factor-α (TNF-α). In addition, reactive oxygen species and proteases such as urokinase-type plasminogen activator (uPA) are produced by neutrophils to fight pathogens and debride devitalized tissue. After a few days, neutrophils begin to undergo cell death and are replaced by macrophages that are either resident or recruited from circulating monocytes in response to proinflammatory cytokines and chemotactic gradients. Activated macrophages are thought to play an integral part in coordinating the wound response by providing matrix remodeling capabilities (uPA, MMP9), synthesis of growth factors (fibroblast growth factor [FGF], PDGF, transforming growth factor beta [TGF-β]), and production of angiogenesis factors (VEGF). These factors activate fibroblasts to synthesize new ECM and promote neovascularization in the formation of granulation tissue. Other cells that are important in wound healing include mesenchymal stem cells.23 These fibroblastoidlike cells can be mobilized from the bone marrow or from niches within various tissues in order to aid wound healing by differentiating into different connective tissue cell types.
Cancer cells are often surrounded by activated fibroblasts and BMDCs. Because of the resemblances between primary tumors and normal tissue wound response, cancer has been described as a “wound that does not heal.” Although Virchow hypothesized in the 1850s that inflammation was the cause of cancer, the presence of an inflammatory response has generally been interpreted as evidence that the immune system actively fights the cancer as it does with invading bacterial or viral pathogens. Under this scenario, the inflammatory response would apply significant selective pressure on the tumor to evade immune-mediated attack, and the nonhealing nature of the response suggests a back-and-forth struggle. Tumors that progress do so by orchestrating an immunosuppressive environment, a process known as immunoediting.24
To facilitate an immunosuppressive environment, the tumor microenvironment selects for cells that favor production of immunomodulatory factors like TGF-β, cycoloxygenase-2 (COX2), CSF-1 (macrophage growth factor, colony-stimulating factor-1), IL-10, and IL-6. These cytokines inhibit maturation of dendritic cells and promote tumor-associated macrophages (TAMs) that are immunosupressed.25 Tumors also recruit BMDCs that have immunosuppressive properties such as myeloid-derived suppressor cells (MDSCs).26 These cells are recruited through signaling events that involve stromal cell-derived factor-1 (SDF-1, also known as CXCL12) and its ligand CXCR4 and CXCL5/CXCR2, another chemokine/receptor pair. On arrival, the MDSCs increase local production of TGF-β, block T-lymphocyte function, and inhibit the activation of NK cells.27 Thus, although the inflammatory response undoubtedly can help to limit cancer growth, cancers seem to select for cells that create immunosuppressive surroundings.
Rather than simply suppress the inflammatory response, cancer cells actually develop mechanisms to both co-opt and perpetuate it. For example, at the same time MDSCs are contributing to immunosuppression, these cells also facilitate tumor invasion by residing at the invasive front and secreting MMPs. The comingling of various stromal cells and other BMDCs with the cancer also actively contributes to tumor growth in a similar fashion. For example, TAMs are often found at points of basement membrane breakdown and, like MDSCs, end up at the invasive front to help tumors degrade extracellular proteins using uPA and MMPs or stimulate tumor growth and motility through EGF receptor ligands and PDGF. As in normal wound healing, growth factors secreted by the TAMs activate fibroblasts. These activated fibroblasts become carcinoma-associated fibroblasts (CAFs) and promote primary tumor growth by secreting CXCL12 to stimulate CXCR4 on tumor cells. Angiogenesis is also aided by the action of CAFs through recruitment of endothelial progenitor cells by CXCL12 and by the action of TAMs that are recruited to areas of hypoxia to produce VEGF. Figure 10.2. presents a summary of this interaction. Thus, although the question of whether the immune system is a friend or foe of malignancies is not a new one, it would seem that recent answers suggest that cancers actually find ways to turn an enemy into an accomplice.
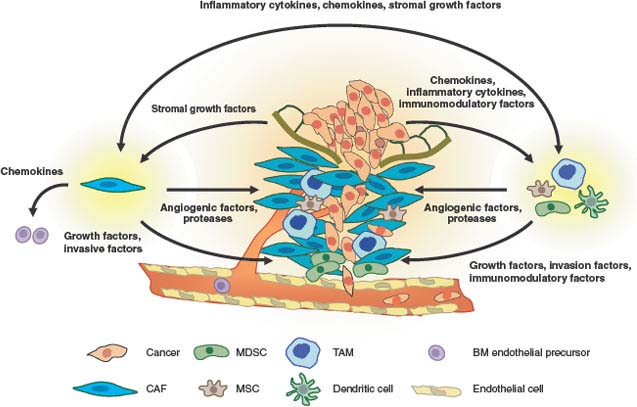
FIGURE 10.2 Interactions between cancer and stroma that promote invasion and metastasis. Cancerized stroma consists of fibroblasts, inflammatory cells, and other bone marrow-derived cells that have been conscripted to aid the tumor in overcoming hypoxia and in invasion and migration. Tissue breakdown, hypoxia, and inflammatory cytokines and chemokines secreted by the tumor cells result in recruitment of tumor-associated macrophages (TAMs), carcinoma-associated fibroblasts (CAFs), mesenchymal stem cells (MSCs), and myeloid-derived suppressor cells (MDSCs). TAMs and MDSCs can be found at points of basement membrane breakdown and at the invasive front of the tumor. These cells produce angiogenic factors to promote vascularization, proteases to degrade the extracellular matrix, and growth factors that stimulate tumor invasion and motility. CAFs also produce similar angiogenic factors, protease, and tumor growth factors. In addition, CAFs recruit bone marrow-derived endothelial precursors for angiogenesis. The cytokines and growth factors that TAMs and CAFs secrete are mutually beneficial to each other as part of an inflammatory/woundlike response. Cancers have been described as “wounds that do not heal.” This chronic state is maintained by immunomodulatory cytokines that suppress immune functions to ensure a protumorigenic environment.
Escaping Apoptosis and Senescence
A major mechanism to safeguard against a breakdown in tissue homeostasis due to cells that stray, become damaged, or spent, is to have these cells commit programmed cell death, or apoptosis.28 This form of cell suicide is genetically regulated and can be triggered by a variety of signal transduction pathways linked to proteins that monitor environmental cues or act as damage sensors. Common cell intrinsic triggers for apoptosis include oncogene activation or tumor suppressor gene loss. For example, the inappropriate activation of c-MYC or the loss of Rb results in programmed cell death that must be countered by overexpression of antiapoptosis genes such as Bcl-2 or loss of proapoptotic regulators like p53.29 Extrinsic triggers for apoptosis include hypoxia, low pH, reactive oxygen species, loss of cell contact, and immune-mediated killing. Members of the TNF-receptor family can act as death receptors that mediate activation of proapoptotic proteases called caspases in response to loss of ECM adhesion30 or after being engaged by immune cells for elimination. Cancer cells invariably ignore these cues, and their ability to resist cell death likely contributes to successful establishment of tumors.
In addition to apoptosis, senescence is another important barrier to cancer. This exit from the pool of proliferating cells results from telomere erosion, oncogene induction, and DNA damage. Similar to some forms of apoptosis, senescence is p53-dependent. Thus, pressure to escape senescence can result in the loss of p53 or mutations in p53.
Self-Renewal Ability
Normal tissues result from the differentiation of precursor cells called stem cells, which are multipotent cells with self-renewal ability. In the adult, mature differentiated cells serve specialized tasks and have limited proliferative potential. However, adult tissue still undergoes turnover and is maintained through the self-renewal and multilineage differentiation of adult stem cells. Examples of this include skin, mucosa, and hematopoietic cells whereby a limited and spatially restricted pool of adult stem cells asymmetrically divides. One daughter cell maintains the stem cell pool by self-renewal, and the other daughter cell starts the process of terminal differentiation for tissue maintenance. The majority of cancers maintain some resemblance to their tissue of origin by virtue of persistent differentiation, albeit in an abnormal way. Thus, many cells in a tumor population may have limited proliferative potential and be incapable of sustained self-renewal, similar to their normal counterparts. The idea that only a limited subset of cells in a cancer is capable of self-renewal is called the cancer stem cell hypothesis.
The existence of cancer stem cells was first demonstrated in acute myeloid leukemia and recently shown in breast cancer, glioblastoma, and other cancer types.31 These studies use cell surface markers to enrich for putative stem cell populations. In the case of breast cancer, CD44high/CD24low cells were found to form tumors when injected into immunocompromised mice in low numbers and give rise to a diverse population that contained additional CD44high/CD24low cells. In contrast, the injection of thousands of cells from other populations was nontumorigenic. Thus, the need for cancer cells to acquire self-renewal ability is paramount to the productive growth of the tumor. In principal, however, there is no advantage for cancer to keep this subpopulation of tumor-initiating cells small or fixed. Thus, tumors may contain a large proportion of cells with a tumor-perpetuating stem phenotype,32 and this phenotype may be subject to back-and-forth reprogramming.
COUPLING TUMORIGENESIS WITH METASTASIS INITIATION
The selective pressures previously described that are encountered during primary tumor growth—hypoxia, inflammation, apoptosis, senescence, and need for proliferative, metabolic, and self-renewal sufficiency—drive primary tumors to acquire tumorigenic alterations that support aggressive growth. These same pressures collaterally support the initial stems of metastasis, and remain important throughout the subsequent malignant steps. One of the most striking types of metastasis initiation functions is EMT.
Selecting for Epithelial-to- Mesenchymal Transition
During development, the generation of many adult tissues and organs results from a series of EMT events and the reverse process, a mesenchymal-to-epithelial transition (MET).33 For example, gastrulation and delamination of the neural crest are early developmental processes that layout the three germ layers (ectoderm, mesoderm, and endoderm) and give rise to diverse cell lineages (craniofacial cartilage and bone, smooth muscles, neurons, melanocytes), respectively. Both of these developmental events are characterized by epithelial cells loosening their cell-cell adhesion, losing cell polarity, and gaining the ability to invade and migrate under controlled cues. Important regulators include Notch and Wnt/β-catenin pathways, TGF-β family members, and FGF proteins that serve to set up regulatory networks involving EMT transcription factors such as Snail and Twist. These networks do not necessarily regulate cell fate, but rather drive morphogenetic movements by repression of the cell-cell adhesion protein E-cadherin, promoting cytoskeletal rearrangement, and increasing MMP activity. After cells complete EMT-mediated morphogenetic migration, they can then transiently differentiate into epithelial structures by repressing Snail and undergoing a MET. An example of cells that undergo a primary EMT that is followed by a MET includes neural crest cells giving rise to somites. These epithelial somites that have already experienced a primary EMT that is followed by a MET can then initiate a secondary EMT, which eventually results in development of muscle cells.
Growing evidence points toward EMT as an important characteristic of metastasis-prone cancers. EMT in cancer is not a concrete and tidy single process but rather a collection of cell reprogramming phenomena that share the property of down-regulating epithelial cell markers and, for convenience, the collective denomination of EMT. Hypoxia can induce Snail and Twist, a direct target of HIF-1α.34 Low oxygen enhances β-catenin activity by inhibiting the activity of glycogen synthase kinase-3β, which normally induces the destruction of β-catenin. Accordingly, the presence of enhanced β-catenin signaling promotes Snail expression and subsequent EMT. Interestingly, the ability of hypoxia to liberate active β-catenin may set in place a feed-forward loop to help maintain EMT. Activation of Snail represses E-cadherin, which can then further enhance β-catenin and reinforce Snail expression.
Similar to hypoxia, the inflammatory microenvironment can also promote EMT. It has recently been demonstrated that TNF-α, which is an inflammatory mediator secreted by TAMs, sets into motion a signaling cascade that funnels through NF-kB and glycogen synthase kinase-3β to stabilize Snail and β-catenin, and thus, enhances cancer cell migration.35 This ability of cancer cells to awaken an EMT program by co-opting an inflammatory microenvironment may be further reinforced by EMT itself. Snail-induced EMT is able to generate an environment of immunosuppressive T-regulatory cells and impaired dendritic cells partly through TGF-β and thrombospondin-1, helping to further perpetuate the inflammatory surroundings.36
Besides hypoxia and inflammation, the need for primary cancers to resist apoptosis and overcome senescence may be additional reasons to flip an EMT switch. Cells that have undergone EMT are associated with increased resistance to apoptosis, possibly through prosurvival activity conferred by Snail and Twist.37,38 EMT can also help cancer overcome oncogene-induced senescence. Both Twist transcription factors and ZEB1 have been shown to suppress p21cip1 and/or p16ink4a, two p53-regulated cell cycle proteins that are critical in restraining oncogene-transformed cells via senescence.39 These findings suggest that the pressure to resist apoptosis and bypass senescence can result in activation of EMT transcriptional regulators.
Acquiring the ability to sustain long-term self-renewal would provide an enormous advantage to cells in a growing tumor mass. Recent data suggest that selection for cells that are capable of undergoing EMT may provide a pool of cells with stemlike features.40 Stem cell-enriched subpopulations with a CD44high/CD24low cell surface marker profile have significantly higher levels of many EMT-related transcription factors, such as Snail and Twist, when compared with their CD44low/CD24high counterparts. Furthermore, these EMT transcription factors are able to directly increase the number of cells with stemlike characteristics. Although it remains to be determined whether EMT is occurring in cancer stem cells or whether EMT occurs in non–self-renewing cells that then give them stemlike properties, these data argue for a another compelling reason for cancer cells to acquire EMT properties.
Epithelial-to-Mesenchymal Transition in Metastasis Initiation
Although the ability to awaken an EMT program would seem to offer numerous advantages for cells trying to expand within a primary tumor, it is not clear that primary tumor growth and invasion requires EMT. In fact, EMT-independent mechanisms have been shown to operate and allow carcinomas to invade and migrate at invasive fronts.41 Despite this, mounting evidence suggests that for cancer cells that do satisfy tumorigenic functions with expression of EMT genes, these cells are also endowed with metastasis initiation functions. Early evidence that EMT genes can be dispensable for primary tumor growth but essential for metastasis has come from analyses of Twist.42 In a breast cancer model, inhibition of Twist was shown to have no effect on primary tumor growth but potently reduced the number of metastatic lesions in the lung. Consistent with these findings, inhibiting Twist in either hypoxic cells or in cells overexpressing HIF-1α reversed both EMT and metastasis,34 and inhibiting Snail decreased metastasis induced by inflammatory signals.35 In total, these observations suggest that when imposing forces like hypoxia and inflammation select for cells capable of EMT, EMT genes become important in metastasis.
What makes EMT cells particularly adept at initiating early metastatic events? A careful analysis of the effect of Twist on metastasis revealed a role for Twist in establishing high levels of circulating tumor cells through enhancing intravasation and/or survival in the circulation.42 The ability of cells undergoing EMT to intravasate is consistent with observations that EMT occurs at the invasive front of tumors whereby cells lose E-cadherin, detach, invade, and break down the basement membrane. Accordingly, experiments that directly analyzed EMT and non-EMT cells showed that only the EMT cells were able to penetrate surrounding stroma and intravasate.43
Another reason that EMT can make cells susceptible to distant spread may be related to the ability of EMT to confer stemlike properties. For the same reasons that primary tumors may rely on cancer stemlike cells for continued self-renewal, metastasis formation may also rely on cancer stemlike cells. Using pancreatic cancer models, it has been shown that a distinct subpopulation of CD133+/CXCR4+ cells localizes to the invasive edge of tumors and is more migratory than CD133+/CXCR4− cells.44 Interestingly, although both populations were equally capable of instigating primary tumor growth, only the CD133+/CXCR4+ cells could metastasize to the liver. Targeting CXCR4 either through depletion or pharmacologic targeting interfered with spontaneous metastasis formation. In breast cancer patients, CD44high/CD24low cells, which are increased by EMT and express EMT markers, display stemlike properties and can be enriched both in the circulation and in the bone marrow.45 Thus, EMT may play a role in metastasis initiation by ensuring that distant outgrowths are maintained with a pool of stemlike cells.
Prevalence of Primary Tumors with Metastasis-Initiation Functions and Early Dissemination
Considering that EMT can be selected by hypoxia and inflammation, resulting in invasive cancer cells that can intravasate and self-renew, one expectation might be that primary tumors can acquire metastasis-initiation functions early during tumorigenesis because EMT is acquired early. Indeed, recent evidence suggests that initial steps in metastasis can frequently occur far earlier than previously thought. By using transgenic mouse models of breast cancer, it has been shown that disseminated tumor cells (DTCs) can be found in bone marrow and lung tissue even before morphologic evidence of primary tumor invasion.46,47 Closer inspection with electron microscopy reveals individual tumor cells from preinvasive atypical hyperplastic lesions breaching the basement membrane. Furthermore, preinvasive lesions also displayed high levels of Twist and MMP expression. These early DTCs progressed in parallel with the primary tumor and could give rise to metastatic lesions. Such experimental observations are consistent with clinical findings that patients with preinvasive DCIS also have cytokeratin-positive tumor cells in the bone marrow, as do a significant fraction of patients with early-stage breast cancers. However, despite these findings, patients with DCIS rarely develop metastasis, which highlights that metastasis-initiation functions are not sufficient to complete the metastatic cascade and indicates the need for metastasis-progression and macrometastatic-colonization genes.
COUPLING TUMORIGENESIS WITH METASTASIS PROGRESSION
The selection of genes that primarily fulfill tumorigenic functions may also result in genes that secondarily aid cancer cells after they have found their way into the circulation. In other words, when genes are selected to help the primary tumor grow, some of these genes have a collateral effect of benefiting the cancer after it disseminates by providing metastasis-specific tools. Such genes can be classified as metastasis-progression genes; however, a clear distinction with metastasis-initiation genes may not always be evident. In this section, we describe functions that can be classified as metastasis-progression functions.
Premetastatic Niche
Even before tumor cells colonize distant organs, they can help prepare foreign soil for the subsequent arrival of DTCs by remotely coordinating a “premetastatic niche” from the primary tumor.48 These niches are often located within distant organs around terminal veins and are characterized by newly recruited hematopoietic progenitor cells of the myeloid lineage and by stromal cells. This niche provides an array of cytokines, growth factors, and adhesion molecules to help support metastatic cells on their arrival.
The remote coordination of the premetastatic niche by the primary tumor appears to be through cytokines and growth factors associated with the inflammatory and hypoxic microenvironment of the primary cancer. Through the secretion of cytokine profiles that include VEGF and placental growth factor, primary tumor cells can direct VEGFR1+ myeloid cells to mobilize from the bone marrow and preferentially localize to areas in target organs with increased fibronectin.49 If the primary tumor secretes VEGF, this promotes fibronectin deposition in the lung and directs the construction of the premetastatic niche there, while the combination of VEGF and placental growth factor leads to a more widespread pattern of niche assembly. Similarly, VEGF, TGF-β, and TNF-α produced at the primary tumor site can signal production of inflammatory proteins like S100A8 and S100A9 specifically within the lung parenchyma.50,51 This results in the infiltration of myeloid cells into the lung and subsequent formation of the niche. Besides inflammatory signals from the primary tumor, LOX produced by a hypoxic primary tumor environment can also direct formation of a premetastatic niche.52
Once myeloid and activated stromal cells form the premetastatic niche, the local environment in the distant organ is altered by the production of inflammatory cytokines and MMPs, which begins bearing an evolving resemblance to the primary site. Consequently, when primary tumor cells start wandering in the circulation, the target organs with an established premetastatic niche become a better soil in which to attach, survive, and grow. Indeed, if the formation of the niche is disrupted, metastasis is inhibited. In this way, the shower of cytokines and growth factors that accompany inflammatory and hypoxic responses at the primary tumor not only selects for cancer cells that can flourish in the primary site but has the secondary effect of creating a more welcoming environment in distant organs after dissemination. Such a scenario would argue that the mechanism of premetastatic niche formation has properties consistent with metastasis-progression functions.
Survival in the Circulation
From experimental model systems, it has been estimated that approximately one million cancer cells per gram of tumor tissue can be introduced daily into the circulation.53 Direct inoculation of tumor cells into mice demonstrates that metastasis can be an inefficient process because despite large numbers of circulating tumor cells (CTCs), relatively few metastases form.8 In humans, the inefficiency of CTCs to give rise to detectable metastases was inadvertently demonstrated in ovarian cancer patients who received peritoneal-venous shunts for palliation of malignant ascites.54 Despite the re-routing of millions of cancer cells from the peritoneum to the venous circulation, for years in some cases, the majority of the patients did not develop widespread metastases. Thus, even if cancer cells acquire metastasis-initiation functions like EMT, the ability to merely enter into the circulation often is not a rate-limiting step in metastasis. Other obstacles must be overcome.
After intravasation into the circulation from the primary tumor, tumor cells encounter significant physical stress due to shear forces or mechanical arrest in small-diameter vessels. The hepatic sinusoids can be activated by the mechanical restriction of tumor cells to secrete nitric oxide. Nitric oxide can cause apoptosis of arrested tumor cells and has been shown to be required for the massive cell death of experimentally injected melanoma cells.55 Endothelial cells can also guard against wandering tumor cells through expression of DARC, a Duffy blood group glycoprotein.56 DARC interacts with KAI1 expressed on circulating tumor cells causing them to undergo senescence. KAI1 was originally identified as a metastasis suppressor gene. The immune system can also actively attack circulating tumor cells.57 For example, NK cells can engage and kill cancer cells via TNF-related molecules such as TRAIL or CD95L. In total, these mechanical and cell-mediated stresses can result in a short half-life for CTCs. Estimations derived from the enumeration of CTCs before and after removal of the primary tumor in patients with localized breast cancer demonstrate that the half-life can be as short as a few hours.58
How can CTCs evade cell death to enhance their metastatic potential? Growth at the primary tumor site will involve a selection for increased resistance to apoptosis. Antiapoptosis genes such as BCL2 or BCL-XL, or the loss of proapoptotic genes and genes downstream of the TNF-related receptor family, can result in increased metastasis.59,60 Part of this may be the result of survival both in the circulation and shortly after extravasation. Both CTCs and platelets can also express the αvβ3 integrin to promote aggregation of these cells to form tumor emoboli.61 This aggregation not only facilitates arrest but can protect against shear forces and NK cell-mediated killing. Activation of αvβ3 can result from CXCL12/CXCR4 signaling and has been shown to be required for formation of tumor emboli and metastasis.62,63 Thus, the ability of a primary tumor to respond to apoptosis, senescence, and inflammatory signals can secondarily make cancer cells better able to survive in the circulation once metastasis has been initiated.
Extravasation and Colonization
After arresting in capillaries, tumor cells that are able to survive can grow intravascularly. This can lead to a physical disruption of the vessels.64 However, more selective processes of extravasation exist. Cancer cells can mimic leukocytes and bind to endothelial E- and P-selectins.65 Molecular mediators of extravasation include the cytoskeletal anchoring protein Ezrin, which links the cell membrane to the actin cytoskeleton and engages the cell with its microenvironment. Ezrin was discovered to promote metastasis in osteosarcoma by preventing cell death during migration into the lung.66 VEGF expression by the tumor can also lead to disruptions in endothelial cell junctions and facilitate extravasation of cancer cells through enhanced vascular permeability. This is likely mediated by the activation of SRC family kinases in the endothelial cells, which is consistent with decreased lung metastasis in SRC nullizygous mice.67 Expression of hypoxia-induced CXCR4 on CTCs allows for the selective extravasation into certain organs. This selectivity is due to the expression of its ligand CXCL12 by certain organs that include the lung, liver, bone, and lymph nodes.68 Thus, the selection to successfully deal with hypoxia during primary tumor growth may bias the pattern of distant spread through the CXCR4-CXCL12 pathway.
The steps of extravasation and establishment of micrometastases help to illustrate a more concrete example of the concept of metastasis-progression genes. Using a mouse model system for breast cancer metastasis, a gene expression signature for aggressive lung metastasis was discovered that not only mediated experimental lung metastasis but was also expressed by primary human breast cancers with increased risk of metastasis selectively to the lung.17 Four members of this lung metastasis gene expression signature (LMS), namely EREG (an EGF receptor ligand), MMP1, MMP2, and COX2, were selected during primary tumor growth and conferred a growth advantage by facilitating the assembly of new blood vessels.16 The vascular remodeling program coordinated by these four genes was also critical to the extravasation of circulating breast cancer cells into the lung parenchyma, as its inhibition resulted in the intravascular entrapment of single cells. Another LMS gene that is a mediator of lung extravasation is the cytokine angiopoietin-like 4 (ANGPTL4).69 ANGPTL4 promotes the dissociation of endothelial cell-cell junctions and, consistent with it being a metastasis-progression gene, ANGPTL4 is induced by TFG-β produced in the primary tumor microenvironment. The combined examples of VEGF, CXCR4, and several LMS genes illustrate how metastasis-progression genes can arise during primary tumor growth through the acquisition of genes that can cope with or respond to selective pressures at the primary site.
Cancer cells that have initiated the metastatic cascade can also epigenetically acquire metastasis-progression functions through paracrine factors secreted by the microenvironment of primary tumors. Mesenchymal stem cells recruited to the inflammatory stroma of a primary tumor have been shown in a xenograft model to produce CCL5 as a result of heterotypic interaction with neighboring breast cancer.70 CCL5 can then act in a paracrine fashion to enhance motility, invasion, and extravasation into the lung. Accordingly, the prometastatic effects mediated by CCL5 were reversible when interactions with MSCs were removed. Thus, metastasis-progression functions acquired through events that occur in the primary tumor need not be accompanied by somatic changes in metastasis-progression genes.
Tumor Self-Seeding
As cancer cells selected by the inflammatory and hypoxic surroundings of the primary tumor wander through the circulation, might the most hospitable and likely destination for extravasation be the primary tumor from which they came? At least in theory, it would seem that compared with uncharted foreign environments or even premetastatic niches, the primary tumor would impose the least resistance to colonization.71 Support for this concept of tumor self-seeding has recently been provided using mouse model systems and a variety of different cancer types by demonstrating that CTCs can seed the primary tumor and contribute to its mass.72 The ability to self-seed is promoted by IL-6 and IL-8, common prometastatic cytokines found in the tumor microenvironment. The cancer cell expression of MMP1 and Fascin-1, two previously identified lung metastasis genes, facilitates transendothelial migration and tumor self-seeding. In fact, metastatic cells in general are more efficient seeders, which may result from a direct cause and effect. Subpopulations of cancer cells selected to be efficient seeders can express an array of multiorgan metastasis genes. These observations suggest that disseminated cells capable of successfully navigating a round-trip rather than a one-way excursion help the primary tumor select for cells with metastatic functions.
FROM METASTASIS PROGRESSION TO MACROMETASTATIC COLONIZATION
Because metastasis-initiation functions and metastasis-progression functions are coupled to tumorigenic functions, the accumulating evidence that primary tumors can exhibit metastatic traits early on during primary tumorigenesis and with such high prevalence may not be surprising. However, despite the ability of EMT to drive invasion, intravasation, and self-renewal, and despite the remote influence the primary tumor has on survival in the circulation, extravasation, and premetastatic niche formation, the completion of the metastatic cascade is still relatively infrequent for many cancer types. This suggests an important requirement for macrometastatic colonization functions, or functions selected at distant sites for organ-specific colonization.
Dormancy
A major limiting step in metastasis is acquiring the ability to sustain growth within a distant site after extravasation. Many cancers such as breast and prostate will not give rise to metastasis until years or even decades after eradication of the primary tumor. Clinically, there is also a clear discrepancy between the proportion of patients with preinvasive or early-stage cancers that have cytokeratin-positive cells in the bone marrow and the proportion that develop overt metastasis. Experimentally, it has been shown that the vast majority of extravasated cancer cells do not form macrometastasis.8 In aggregate, these observations of latency are referred to as metastatic dormancy and point toward the bottleneck that forms prior to outgrowth of macrometastasis.
A distinction has been made between tumor mass dormancy, which describes the process that inhibits expansion of a dividing tumor population, and cellular dormancy, which describes when cancer cells enter a state of growth arrest.73 Tumor mass dormancy has been attributed to the existence of preangiogenic micrometastasis in which cell division is balanced by apoptosis.74 Clinical evidence for this comes from one study whereby breast cancer patients with no evidence of disease years after mastectomy and considered candidates for having metastatic dormancy were shown to have detectable CTCs.58 Later acquisition of angiogenic properties may allow such micrometastases to become vascularized and emerge from their occult state. Work with experimental models suggests that indolent metastasis may also be activated by coexisting with more aggressive metastasis secrete systemic factors like osteopontin (OPN).75 Mechanisms that contribute to cellular dormancy may relate to the balance between the RAF-MEK-ERK pathway and the p38 MAPK pathway. Inhibition of the former and activation of the latter is associated with cellular quiescence in a G0-G1 state, and the exact balance between the two may depend on cross-talk between the tumor and the new microenvironment. Genes that may be important in blocking productive cross-talk between dormant metastasis and its microenvironment include metastasis suppressor genes such as NME23, MKK4, and RKIP, to name a few.73,76 This functional class of genes specifically prevents growth of metastasis without influencing primary tumor growth.
Regardless of the nature of the dormancy, dormant cells must acquire genetic or epigenetic changes during residency at foreign sites in order to evolve into gross metastases. Tumor cells that are detected in the bone marrow and likely disseminated from the primary tumor at an early stage during tumorigenesis have been shown to undergo a separate evolution under separate selective pressures. Comparative genomic hybridizations of single breast cancer cells isolated from bone marrow were shown to be genetically heterogeneous in patients without overt metastasis.77,78 These cells also showed few genetic features in common with matched primary tumors. In contrast, bone marrow-derived breast cancer cells from patients with overt metastatic disease displayed a marked reduction in genetic heterogeneity compared with patients with occult disease. These results are consistent with disseminated tumor cells departing from the primary tumor early on and being driven by different selective pressures found in the distant organ. Different metastatic sites that host dormant cells likely contribute unique selective pressures. This paves the way for the acquisition of macrometastatic colonization functions.
Organ Selective Growth
Bone
Homeostasis of the bone is maintained by a constant state of remodeling such that no net gain or loss of bone occurs. The mineralized bone matrix is reabsorbed by osteoclasts and filled in by osteoblasts. The differentiation of osteoclasts from bone marrow mononuclear cells is controlled by CSF-1 and the RANK receptor (reviewed in refs. 79 and 80). RANK interacts with its ligand RANKL produced by osteoblasts, leading to a tight coupling of these two cells with opposing actions. The activity of RANKL can also be controlled by osteoprotegerin (OPG). OPG is a secreted antagonist of RANKL and prevents interaction with RANK and resulting osteoclastogenesis. The differentiation of osteoblasts results from bone marrow mesenchymal stem cells under the control of a variety of regulators including insulin-like growth factor (IGF), endothelin-1, bone morphogenetic proteins, and WNT proteins.
The bone is one of the most common sites of distant spread. However, the latency period that precedes the development of gross osseous metastasis can be years, as illustrated by the high proportion of breast cancer patients with dormant cancer cells in the bone marrow. One factor that provides the ability to sustain this latency period is the SRC kinase, which is required for CXCL12- and IGF-mediated survival signals that protect indolent breast cancer cells in the bone marrow from TRAIL-mediated apoptosis.81 Presumably, the sustained survival and dormancy afforded by SRC would allow time for the acquisition of bone-specific macrometastatic colonization genes.
When the dormancy period expires, gross osseous metastases are characterized by two basic types: osteoblastic and osteolytic. Many tumors such as lung, kidney, and breast carcinomas typically produce osteolytic lesions. Breast cancer cells achieve osteolytic metastasis competency by secreting factors including parathyroid hormone-related protein (PTHrP), TNF-α, IL-1, IL-6, IL-8, and IL-11 in order to orchestrate a vicious cycle of enhanced osteoclast activation, degradation of bone matrix, and the release of matrix-associated cytokines that stimulate the cancer cells79,80 (Fig. 10.3). The secretion of PTHrP leads to the production of the membrane-bound RANKL on osteoblasts, resulting in RANK-mediated osteoclast activation. Other proinflammatory cytokines such as TNF-α, IL-1, IL-6, and IL-11 that are secreted by the tumor cell can lead to a synergistic effect on RANK-mediated signaling and may also inhibit production of OPG. Interestingly, RANK is also expressed on cancer cells and stimulates migratory activity.82 On degradation of the bone matrix, embedded growth factors are released, including TGF-β and IGF. These liberated growth factors can further stimulate the tumor cells and enhance the entire vicious cycle.
A genomewide screen for genes involved in breast cancer bone metastasis uncovered IL-11, MMP1, ADAMTS1, CXCR4, connective tissue growth factor (CTGF), and OPN as mediators of osteolytic bone metastasis.83 CXCR4 may enhance colonization of breast cancer cells to bone, MMP1 and ADAMTS1 proteolytically activate EGF receptor ligands,84 and OPN and IL-11 may promote osteoclast differentiation. The expression of IL-11, CTGF, and PTHrP in breast cancer cells are targets for TGF-β, which is released by the bone matrix.85 In total, these experimental data illustrate how tumor cells can survive and grow in the foreign environment of the bone by coercing resident cells to release secreted factors that are advantageous to the tumor. This cooperative situation between tumor cells and bone marrow cells is similar to that in the primary tumor whereby BMDCs and stromal cells act as accomplices.
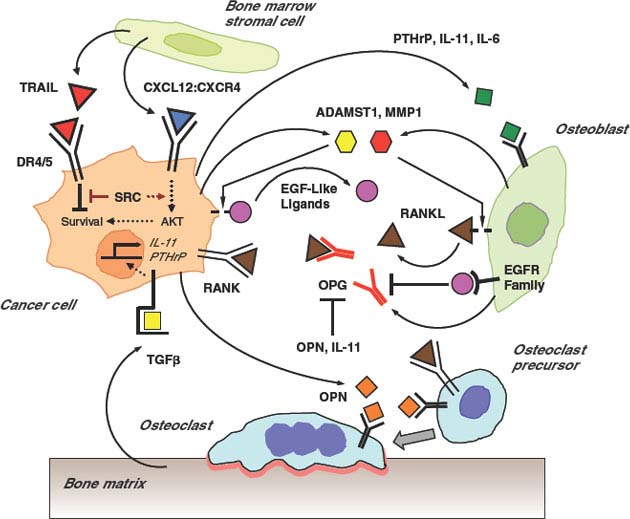
FIGURE 10.3 Interactions between cancer and the bone microenvironment lead to a “vicious cycle.” Cancer cells can migrate to the bone microenvironment under the influence of CXCL12 (blue triangle), which also leads to cell activation and migration via its receptor CXCR4. The CXCL12:CXCR4 interaction promotes survival of metastatic cancer cells through AKT and SRC by counteracting proapoptotic stimuli directed by TRAIL (red triangles) and its receptor DR4/5. Survival can lead to dormancy, utilization of acquired metastasis-progression functions, and opportunities to accumulate additional macrometastatic-colonization genes. Surviving metastatic cells secretes factors such as PTHrP, IL-11, and IL-6 (green squares) that lead to production of membrane-bound RANKL (brown triangles) on osteoblasts. Tumor cell secretion of MMP1 (red hexagon) and ADAMST1 (yellow hexagon) can mobilize RANKL from osteoblasts and EGF-like ligands (magenta circles) from tumor cells. RANKL:RANK interaction stimulates tumor cells and promotes osteoclast differentiation and activation. RANKL can be antagonized by the decoy receptor osteoprotegerin, or OPG (red-colored receptor), that is normally produced by osteoblasts. Engagement of EGF receptor family members can inhibit OPG expression and thereby promote osteoclast activation and metastatic growth. Other tumor-derived factors like osteopontin, or OPN (orange squares), also promote osteoclast activation. Activation of osteoclasts leads to degradation of the bone matrix and release of TGF-β (yellow square). This further stimulates the tumor cells to transcriptionally activate genes such as IL-11 and PTHrP, which contributes to the vicious cycle.
Unlike osteolytic bone metastasis, osteoblastic metastasis is less common and typified by prostate cancer. Osteoblastic lesions result from the preferential stimulation of osteoblasts and/or the inhibition of osteoclasts. Like breast and other cancers that cause osteolytic lesions, prostate cancer also hijacks homeostatic regulation of bone remodeling to its advantage (reviewed in ref. 86). Various paracrine factors secreted by prostate cancer cells can regulate osteoblast proliferation or differentiation, including bone morphogenetic proteins, WNT, TGF-β, IGF, PDGF, FGF, and VEGF. Many of these signals converge on activation of RUNX2, a transcription factor that is essential for bone formation. In turn, the osteoblasts produce factors that stimulate the proliferation of prostate cancer as demonstrated by coculture experiments with either osteoblasts or their conditioned media. The exact factors that drive progression of prostate cancer metastasis have not been firmly established. Nonetheless, these results support the paradigm that a reciprocal relationship exists between the cancer cell and the cells of the microenvironment that contribute to bone-selective metastasis.
Lung
Lung is a common site of metastasis for many different cancers. Experimentally, colonization and growth in animal lungs has been used extensively to study metastasis and invasion by the direct inoculation of tumor cells into the venous circulation. With this method, the lung is the first organ encountered and may be the only organ encountered because of entrapment of tumor cells by the lung capillary bed. Although the list of genes that can contribute to enhanced experimental lung metastasis studied in this way is extensive, it can be difficult to know whether the genes are regulating a generic invasion pathway or contributing to organ-specific growth.
In a study seeking to gain insight into organ-selective metastasis genes, single cell-derived clones from a human breast cancer cell line were discovered to exhibit varying degrees of metastatic ability to the bone and to the lung.87 Although there was no correlation between clones that metastasized well to the lung and those that metastasized effectively to the bone, metastatic tissue tropism did correlate with similarities in global gene expression patterns among the clones. These observations lead to the discovery of an LMS.17 Several of these genes (some have already been discussed) were shown to cooperatively mediate experimental lung metastasis and consisted of secreted factors (EREG, CXCL1, ANGPTL4, and SPARC), cell surface receptors (VCAM1 and IL13Rα2), extracellular matrix protein (TNC) and proteases (MMP1 and MMP2), and intracellular effectors (ID1, Fascin-1, and COX2). Although these genes mediated lung metastasis, they did not enhance bone metastasis and were largely distinct from a previously defined bone metastasis gene expression signature from the same cell line.
Many of the LMS genes are also expressed in a subset of primary human breast cancers. These patients with LMS-expressing tumors are at a higher risk for lung metastasis but not metastasis to bone or various other visceral sites.17,88 In addition, LMS-expressing tumors are larger at the time of diagnosis compared with LMS-negative tumors, which is consistent with experimental data showing that LMS genes were selected for during growth in the mouse mammary gland. Mechanistically, the coupling of tumorigenic function and metastatic function is provided by the LMS genes EREG, COX2, MMP1, and MMP2. As described already, these genes are prototypical metastasis-progression genes in that they assist the primary tumor by promoting the assembly of new blood vessels.16 However, as part of the metastatic cascade, these genes and other LMS genes enhance lung metastasis by promoting extravasation and by priming departing cancer cells for the lung parenchyma.69,89 Some of the LMS genes such as IL13Rα2 and SPARC do not enhance primary tumor growth and are not among the LMS genes expressed by primary tumors that track with lung metastasis; however, these genes do promote lung metastasis, making them macrometastatic-colonization genes that may be selected during residence away from the primary site.
Liver
The liver is supplied by both the portal and the systemic circulations. Metastasis to the liver is commonly seen with colorectal cancer and this bias is likely favored by the portal circulation for this organ. Metastasis from other cancers such as melanoma and breast and lung carcinomas generally occurs via the systemic circulation. Besides the bias for liver metastasis from the portal circulation, there is evidence that the microenvironment of the liver may be particularly favorable for metastasis from gastrointestinal tumors. Extracellular matrix extracted from primary rat hepatocytes was found to stimulate colorectal cancer cell lines better than ECM from fibroblasts.90 Furthermore, CAFs isolated from metastatic colon cancer to the liver were found to secrete factors that enhance proliferation of colon cancer to a greater extent than the conditioned media from fibroblasts isolated from uninvolved liver or skin.91 DNA microarray analysis demonstrated that the CAFs were more genetically similar than uninvolved fibroblasts and preferentially expressed genes associated in ECM remodeling (proteoglycan 1), proteases/protease inhibitors (tissue plasminogen activator, tissue plasminogen activator inhibitor type 1), growth factors (PDGF, FGF, CTGF, VEGF, TGF-β2), cytokines (IL-6, MCP-1), and intracellular mediators like COX2. This alteration in the microenvironment as a potential contributor to liver metastasis was also demonstrated for hepatocellular carcinoma, which tends to recur with intrahepatic metastasis. By comparing noninvolved hepatic tissue from patients with and without metastasis, genes associated with inflammation and/or immune responses were found to be associated with a “metastasis-inclined microenvironment.” A major difference was noted in the cytokine profile of the metastasis-inclined tissue, which was strongly biases to a TH2 rather than a TH1 cytokine profile.92 The latter is associated with cytotoxic T-cell activity and the former is associated with a humoral immune response. These different cytokine milieus may be driven in part by high levels of CSF-1.
Besides local microenvironmental influences on liver metastasis, contributing signaling molecules expressed by cancer cells have also been implicated. In a screen for genes associated with colon cancer metastasis to the liver, the PRL-3 tyrosine phosphatase was identified.93 Experimentally, PRL-3 can trigger angiogenesis and enhance invasion. Clinically, its expression is found in primary colorectal tumors and correlates with metastasis. It is unclear whether PRL-3 selectively mediates metastasis to the liver, as its expression is also found in other distant organs to which colorectal cancer relapses.94
Brain
The principal sources of brain metastasis are lung and breast carcinomas. Melanoma, colorectal, and renal cell carcinomas also can relapse in the brain, whereas some cancer types like prostate cancer rarely do. The microenvironment of the brain is unique in the sense that vascular access is more restricted because of the blood–brain barrier. This barrier is composed of tightly adjoined endothelial cells that are lined by basal lamina and astrocyte foot processes. The blood–brain barrier is a special consideration for pharmacologic intervention, but to what extent it uniquely deters circulating tumor cells from colonizing the brain is unclear. Once in the parenchyma, microenvironmental interactions occur with glial cells. Coculture experiments demonstrate enhanced adhesion and growth when astrocytes are partnered with cell lines from brain metastasis compared with lung metastasis.95 This may involve IL-6, TGF-β, and IGF-1. Melanoma cells that metastasize to the brain have high STAT3 transcriptional activity compared with cutaneous metastases or primary melanoma specimens.96 Manipulation of STAT3 activity was able to alter brain metastasis in animal models. Altered STAT3 affects expression of basic FGF, VEGF, and MMP2, and influences melanoma angiogenesis and invasion. Because of the dearth of cross-comparison studies with other organs, the selectivity of these effects for brain metastasis is unclear. Indeed, STAT3 has also been associated with metastasis to other visceral organs.97
In order to identify specific mediators of brain metastasis, a mouse model for breast cancer was used that incorporated both established cell lines and patient-derived samples.98 Several genes were found to mediate metastasis to the brain, including COX2, the EGFR ligand HBEGF, and the α2,6-sialyltransferase, ST6GALNAC5. Notably, some of the newly identified brain metastasis genes overlapped with previously identified lung metastasis genes, a finding that is consistent with the known propensity for patients to develop synchronous or metachronous metastases to both organs. Accordingly, passage through the nonfenestrated capillaries of the brain and lungs could be promoted by COX2 and HBEGF. In contrast, the specificity of the gene signature for the brain could be attributed to ST6GALNAC5 and other associated genes that normally have a brain-restricted expression pattern. ST6GALNAC5 enhances the ability of breast cancer cells to pass through the blood–brain barrier, although the exact mechanism remains unknown. Patients who express the genes in the brain metastasis signature also have a higher risk of succumbing to brain metastasis.
Lymphatics
The physiological function of the lymphatic system is to collect extravasated fluid, proteins, and immune cells from draining organs in order to return them for transport by the circulation. Lymphatics are low shear force vessels composed of a single layer of endothelial cells with little or no basement membrane and sparsely coated with pericytes. Lymphangiogenesis involves VEGF-C, VEGF-D, and their receptor VEGFR-3 (reviewed in ref. 99). In contrast to angiogenesis mediators like VEGF-A that responds to both inflammation and hypoxia, VEGF-C/D expression is induced by inflammation but not hypoxia. For example, macrophages that respond to inflammatory signals are a rich source of VEGF-C/D. Thus, angiogenesis can occur without lymphangiogenesis but under most circumstances lymphatic vessels grow concomitantly with blood vessels. Other non-VEGF family members can also induce lymphangiogenesis and include FGF2, PDGF, and angiopoietin proteins.
Unlike with angiogenesis, the functional advantage for tumors that induce lymphangiogenesis is a matter of debate. Possibilities include lowering interstitial fluid pressure in the tumor to facilitate blood perfusion and combat hypoxia, or the growth of new lymphatics may facilitate mechanisms that contribute to angiogenesis.100 Alternatively, lymphangiogenesis may coincidentally result from induction of angiogenesis and/or the involvement of immune cells. Part of this uncertainty results from questions regarding the degree to which intratumoral lymphatics are functional. Lymphatics can be found in a peritumoral location or reside intratumorally. Although proliferating intratumoral lymphatic vessels are observed in animal models and cancers of the head and neck and melanoma, they may collapse under high intratumoral pressure. At least one study revealed that these intratumoral lymphatics were not important in conducting metastasis, rather peritumoral lymphatics were the main route.101 Nonetheless, mouse models have revealed that inducing lymphangiogenesis does lead to lymph node metastasis.102,103 Once in the lymph nodes, lymphatic stromal cells are a source for EGF, IGF-1, and various chemokines.104 Like the lung parenchyma and the bone marrow, lymph nodes secrete CXCL12, which can interact with tumor-expressing CXCR4.68 Other chemokine receptors such as CXCR3 also play a role in lymph node metatasis.105
Metastasis to the regional lymph nodes is considered one of the early signs of metastatic potential and/or distant spread. A long-standing question has been whether lymphatic metastasis selects cells that have enhanced metastatic ability but has not yet spread to distant organs or whether lymph node involvement is a marker for a tumor that may have already become metastatic in general. Reasons for the former include the idea that some tumors can only intravasate into lymphatics but not directly into blood vessels because they cannot overcome higher molecular or physical barriers. Explanations for the latter include underlying molecular mechanisms that couple angiogenesis, lymphangiogenesis, and metastatic ability, resulting in synchronous dispersal. To distinguish between these possibilities, lymphangiogenesis was inhibited in a mouse model for lung cancer metastasis using a soluble VEGFR-3-immunoglobulin fusion protein that traps VEGF-C/D and inhibits VEGFR-3 signaling. This approach blocked lymph node metastasis but had no effect on lung metastasis.106 However, in another study, blocking VEGFR-3 signaling with mammary tumors suppressed metastases to both lymph nodes and the lungs.107 Most likely, lymphatic-dependent and -independent metastasis are both possible and dictated by underlying biological mediators and selective pressures. This would also most fit clinical data. Markers for lymphatic-dependent and -independent spread would be useful to guide whether regional lymph nodes should be addressed therapeutically or less aggressively for prognostic staging.
Early, Multiorgan Metastasis
Most cancers such as breast, prostate, and sarcoma, to name a few, demonstrate appreciable latency periods. Depending on the onset of dissemination and colonization within distant organs, this provides varying and often lengthy periods for acquiring macrometastatic-colonization genes. In contrast, some cancers, like adenocarcinomas of the lung and pancreas, have short latency periods and exhibit early systemic metastasis often to multiple organs. One explanation for short versus long periods of latency is related to origin of the cell population initially targeted for transforming and tumorigenic events. The cancer stem cell hypothesis and the role of EMT in metastasis already suggest that early progenitor cells or cells that may have played a role in developmental processes can be predisposed to activate metastasis-progression mechanisms. An example of this has been demonstrated by introducing defined oncogenic alterations into different cell types.108 When mammary epithelial cells or fibroblasts were transformed, these cells were tumorigenic but not metastatic. In contrast, oncogenic transformation of melanocytes resulted in aggressive metastasis. This difference was because of the expression of the EMT transcription factor Slug in the melanocytes but not the other tumor types. Thus, the transformation of certain unique cell types may predispose to early metastatic behavior and could explain certain phenomenon such as cancers of unknown primary.
Identification of genes that promote early metastasis in adenocarcinoma of the lung also supports the view that some carcinomas may not have an extensive requirement for macrometastatic-colonization functions. By using mouse models to dissect the molecular basis for lung metastasis, the existence of a WNT/TCF pathway was found capable of mediating rapid multiorgan metastasis through LEF1 and HOXB9.109 A clinical predictor based on this pathway was associated with metastatic risk for patients with lung, but not breast, adenocarcinomas. This pathway is well characterized in stem cell and developmental biology, including EMT. Recent findings revealing that important drivers of aggressive metastasis can include developmental-related genes capable of causing genomewide changes110,111 will likely help to uncover additional scenarios whereby macrometastatic-colonization functions play limited roles.
MICRO-RNAs AND METASTASIS
It was once thought that the effects of genes necessarily resulted from the proteins that they encode. However, as a result of developmental studies on the worm Caenorhabditis elegans that identified important roles for noncoding RNAs (ncRNAs), it is now appreciated that ncRNAs potentially influence cellular and organismal phenotypes on par with traditional protein-coding genes. In fact, microRNAs, which are approximately 22 nucleotide-long small ncRNAs, have been discovered to have important effects on metastasis.112 MicroRNAs function by regulating the expression of hundreds of target genes through sequence-specific binding between the microRNA and the 3’ untranslated regions of mRNAs. This engagement results in either the degradation of the target mRNA or the inhibition of protein translation. Through this action, microRNAs can influence a large number of genes. In fact, it is predicted that microRNAs preferentially interact with genes that are central to highly connected networks.113
MicroRNAs can function both as metastasis activators and metastasis suppressors by influencing numerous genes involved in metastasis initiation, progression, and colonization. Several microRNAs, such as miR-10b, miR-9, and members of the miR-200 family, play critical roles in EMT or EMT-related events. For example, miR-10b influences cell migration, invasion, and metastasis by repressing HOXD10, a known inhibitor of a RHOC-mediated promotility program.114 The expression of miR-10b is under the control of the EMT transcription factor Twist. In fact, miR-10b is essential for Twist-induced EMT and for metastasis, making miR-10b a metastasis activator. MiR-9 promotes metastasis by targeting E-cadherin, which results in β-catenin signaling and VEGF expression.115 Accordingly, motility, invasion, angiogenesis, and metastasis ensue. MiR-200 family members also regulate EMT by inhibiting ZEB1 and ZEB2, which normally promote EMT through the suppression of E-cadherin.116–118 Thus, in contrast to miR-10b, miR-200 family members act as metastasis suppressors. Other microRNAs such as miR-335, miR-126, miR-31, and let-7 also suppress distant spread. MiR-335 inhibits metastasis and invasion by targeting the LMS genes SOX4 and TNC, while miR-125 suppresses overall tumor growth and proliferation.119 MiR-31 represses multiple steps in the metastatic cascade, an effect that is related to its influence on multiple metastasis genes.120 Let-7, which is one of the most extensively studied microRNAs, interferes with both self-renewal and distant colonization, and its expression can be controlled by metastasis suppressor genes.76,121 Numerous other microRNAs that play a role in metastasis are being rapidly identified. Interestingly, even non-microRNA ncRNAs are being identified to influence aggressive distant spread.110 The potential for microRNAs to serve as clinical markers and/or therapeutic targets is already starting to show promise.
Selected References
The full list of references for this chapter appears in the online version.
7. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 2003; 3:453.
8. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002;2:563.
9. Hellman S. Karnofsky memorial lecture: natural history of small breast cancers. J Clin Oncol 1994;12:2229.
13. Bernards R, Weinberg RA. A progression puzzle. Nature 2002;418:823.
14. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 2009;9:274.
16. Gupta GP, Nguyen DX, Chiang AC, et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007;446:765.
17. Minn AJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung. Nature 2005;436:518.
25. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010;141:39.
26. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer 2009;9:239.
27. Yang L, Huang J, Ren X, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008;13:23.
33. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139:871.
34. Yang MH, Wu MZ, Chiou SH, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol 2008;10:295.
35. Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009;15:416.
36. Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009;15:195.
40. Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704.
42. Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004;117:927.
44. Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007;1:313.
45. Pantel K, Brakenhoff RH, Brandt B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer 2008;8:329.
46. Husemann Y, Geigl JB, Schubert F, et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008; 13:58.
47. Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science 2008;321:1841.
48. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer 2009;9:285.
49. Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005;438:820.
51. Hiratsuka S, Watanabe A, Sakurai Y, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol 2008;10:1349.
52. Erler JT, Bennewith KL, Cox TR, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009;15:35.
56. Bandyopadhyay S, Zhan R, Chaudhuri A, et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat Med 2006; 12:933.
63. Felding-Habermann B, O’Toole TE, Smith JW, et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci U S A 2001;98:1853.
68. Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001;410:50.
70. Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007;449:557.
71. Norton L, Massague J. Is cancer a disease of self-seeding? Nat Med 2006;12:875.
72. Kim MY, Oskarsson T, Acharyya S, et al. Tumor self-seeding by circulating cancer cells. Cell 2009;139:1315.
73. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 2007;7:834.
75. McAllister SS, Gifford AM, Greiner AL, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 2008;133:994.
78. Schmidt-Kittler O, Ragg T, Daskalakis A, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A 2003;100:7737.
81. Zhang XH, Wang Q, Gerald W, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 2009;16:67.
83. Kang Y, Siegel PM, Shu W, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003;3:537.
87. Minn AJ, Kang Y, Serganova I, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest 2005;115:44.
88. Minn AJ, Gupta GP, Padua D, et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc Natl Acad Sci U S A 2007;104:6740.
89. Gupta GP, Perk J, Acharyya S, et al. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci U S A 2007;104:19506.
98. Bos PD, Zhang XH, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009;459:1005.
101. Wong SY, Haack H, Crowley D, Barry M, Bronson RT, Hynes RO. Tumor-secreted vascular endothelial growth factor-C is necessary for prostate cancer lymphangiogenesis, but lymphangiogenesis is unnecessary for lymph node metastasis. Cancer Res 2005;65:9789.
102. Skobe M, Hawighorst T, Jackson DG, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med 2001;7:192.
103. Stacker SA, Caesar C, Baldwin ME, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med 2001;7:186.
108. Gupta PB, Kuperwasser C, Brunet JP, et al. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet2005; 37:1047.
109. Nguyen DX, Chiang AC, Zhang XH, et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 2009;138:51.
110. Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010;464:1071.
114. Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007;449:682.
116. Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008;10:593.
117. Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 2008;283:14910.
118. Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 2008;22:894.
119. Tavazoie SF, Alarcón C, Oskarsson T, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 2008;451:147.
Related posts:

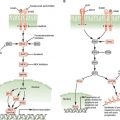
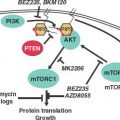

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


