The anatomic structures in which breast cancer invasion occurs arise as paired mammary glands developing from epithelial thickenings on the ventral surface of the 5-week fetus. A mesenchymal condensation occurs around burgeoning epithelial stalks around the 15th week. A physiologic “invasion” of this mesenchyme by cords of epithelial cells creates columns, which later give rise to the lobular organization of the mammary gland. With the onset of cyclical hormonal secretion at puberty, the hormonally responsive periductal stroma differentiates, and the epithelial columns develop into elongated lactiferous ducts terminating into terminal ducts that give rise to lobuloalveolar structures responsible for breast milk production. The adult breast consists of about 15 to 25 lobes, each associated with a major lactiferous duct terminating in the nipple. The glandular and ductal structures are embedded in specialized, hormone-responsive stromal tissue, which consists of adipose tissue admixed with collagenous and vascular elements, the relative abundance of which is largely responsible for the physical and radiographic appearance of the breast (Fig. 2-1).
Figure 2-1
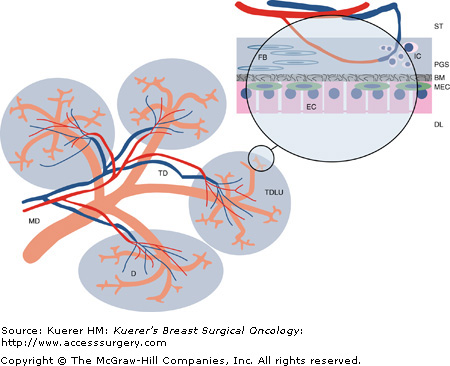
Diagram of breast lobular architecture. This is a stylized depiction of one of the 15 to 25 lobes, or segments, of the human breast. The duct architecture is drawn in orange, illustrating the tree like branching of the ducts into the periphery of the gland. The stromal tissue (ST) is indicated as blank space, and contains the supporting vasculature, of which only the blood vessels are drawn. The gray areas surrounding the terminal duct/lobular units (TDLU) represent the specialized, more dense stromal tissues surrounding the ductal system.
The insert shows a magnified view of a ductule (D) and its lumen (DL), with a layer of epithelial cells (EC), which are connected by intercellular tight junctions. Interspersed between the epithelium and the delimiting basement membrane (BM) are myoepithelial cells (MEC). The periglandular stroma (PGS), containing specialized myofibroblasts (FB), also known as delimiting fibroblasts, commonly harbors inflammatory cell (IC) infiltrates.
There are two distinct cell types forming the epithelium of the lactiferous ducts. The majority are columnar cells lining the surface, which are interspersed with basal cells. Basal cells are thought to give rise to both columnar cells and myoepithelial cells, which separate the epithelial layer from the basal lamina in larger ducts, becoming sparser and discontinuous in the smallest branches and the lobular glands. Throughout the duct system, including terminal ductules and acini, the epithelial and myoepithelial layers are invested by a basal lamina and surrounded by delimiting fibroblasts, separating them from stromal elements of the breast.
Invasion is the second, and arguably the most consequential, of three pivotal milestones in the evolution of breast cancer. Invasion is preceded by the molecular events initiating carcinogenesis, and it may be followed by metastasis, the seeding of cancer cells to foci with no contiguous connection to the primary tumor. Before invasion occurs, the malignant cells, which may be morphologically and biochemically very similar to invasive and metastatic cells, represent a contained, potential threat, which may be completely averted by therapeutic intervention. Significantly, as long as the Rubicon of epithelial organs, the basement membrane, has not been breached, alea non jacta est, as Caesar may have put it, and there can be certainty of cure if the local disease can be completely dealt with. This avoids the ambiguity of probabilistic risk assessments that are the usual mainstay of surgical oncology practice.
This is reflected in the histopathologic terms describing the stages of breast cancer progression, with nonproliferative and proliferative benign breast lesions, atypical hyperplasia, and finally carcinoma in situ, all reserved for cases lacking evidence of any penetration of the basement membranes surrounding these lesions. A breach is acknowledged with the term invasive breast cancer. Since the latter is necessary for access to lymphatic and blood vessels, before which metastatic breast cancer cannot occur, invasive disease is assumed as soon as a metastasis is detected, even if the actual focus has not been identified.
Unsurprisingly, much attention has been given to improve the correct classification of lesions where the determination of invasiveness is difficult, which has given rise to the term microinvasive carcinoma, for cases with an invasive focus of less than 0.1 cm. This category should be distinguished from minimally invasive carcinoma, which refers to unambiguously invasive cancers less than 1.0 cm in diameter.
Biologically, penetrating the basement membrane puts the cancer cells into contact with an entirely new microenvironment. Carcinoma in situ is devoid of vasculature, and cell interactions are limited to cells of epithelial origin. Burgeoning cells in the center of a duct or lobule may die off when distance to the basement membrane exceeds the diffusion limits for nutrient and gas exchange.
Once the basement membrane has been penetrated, cancer cells gain access to the periglandular stroma, with its entirely new set of cell types and molecules. This paves the way to fundamental intracellular changes induced by intense interactions with the stromal cells, growth factors, and the immune system. Vitally, the ability to induce angiogenesis, with its promise of virtually limitless nutrients and oxygen, enables potentially permanent proliferation. The organizing principle of an epithelium, the vertical stratification controlled by cells directly attached to the basement membrane, is lost.
As epithelial tumors progress through progressive stages of carcinogenesis, from benign proliferative states to in situ and invasive cancer, they undergo profound architectural, cell biological, and molecular changes that have been summarized as an epithelial to mesenchymal transition (EMT).1-3 Originally described in the context of normal embryologic development,4 where cells from the ectodermal germ layer can detach and migrate over considerable distances to participate in the formation of various organs, EMT is now understood as a consequence of a complex set of molecular changes that allow the cancer cells to escape the structural constraints of the epithelial architecture, such as the specialized circumferential cell-to-cell adhesion complexes. These include the loss of several epithelial characteristics, such as apicobasal polarity, a term describing the marked differences in cell surface specializations at the top and bottom of an epithelial cell. Changes in cell shape from cuboidal to more spindle-shaped are associated with a reorganization of the actin cytoskeleton; and a decrease in epithelial-specific gene expression, including E-cadherin5,6 and claudins,7 is matched by an increase in mesenchymal gene expression, such as vimentin8 and numerous proteases.
Fundamentally, the essential features of EMT are the disruption of intercellular, epithelial-to-epithelial contacts, and the enhancement of cell motility, which, coupled with the production of extracellular matrix-degrading enzymes, allows the escape from the confines of the breast duct and invasion into the surrounding stroma.
It should be understood, however, that the concept of EMT remains, at this point, largely an attractive but unproven concept that is still vigorously debated.9,10 Much of the controversy revolves around the proposition that EMT necessarily implies a complete transition from an epithelial cell to a mesenchymal cell such as a fibroblast, something that has not been demonstrated in clinical tissues. A complete loss of epithelial characteristics is not observable in pathologic specimens of invasive or even metastatic breast cancer, and the appearance of abnormal, mesenchymal gene expression patterns and markers in cancer cells is not sufficient to reclassify the cells to a different lineage, since such events are a common byproduct of the genetic and epigenetic disarray in cancer cells. Indeed, most of the data supporting the existence of EMT are derived from embryologic and in vitro cancer cell-line model systems, which certainly do not perfectly mimic the in vivo situation. Nevertheless, EMT as intellectual construct to help understand and investigate these processes has been very productive, and there is little reason to abandon the model if one does not assume a complete conversion of one cell type to another.11 Interestingly, recent evidence from a transgenic mouse model using cell-fate mapping strategies does support the possibility of true EMT, although this only occurred in association with c-MYC amplification, and a concurrent microsatellite marker analysis of human tumors suggests this to be a rare occurrence.12
At the level of the cancer cell, EMT is characterized by loss of intercellular junctions and cytoskeletal rearrangements favoring motility. Cell–cell interactions typical of stationary epithelial cells are replaced by cell–matrix interactions favoring migration and invasion, which is further enabled by reciprocal interactions with peritumoral stroma leading to an increased production of matrix-degrading proteases. The ensuing degradation and remodeling of the extracellular matrix (ECM), beginning with the basement membrane, is coupled with proliferative stimuli promoting tumor growth, recreating strands of cohesive multicellular columns. The regaining of a cohesive epithelial appearance has been termed mesenchymal–epithelial transition (MET), and is also an important feature in the establishment of metastatic disease.3,11 It should be noted that this dynamic process leads to significant localized differences in cell function, morphology, and organization. Often, a characteristic invasive front at the advancing edge of the tumor strands can be discerned from more central parts of the tumor, at least in tumors showing more solid morphologies. This is less apparent in diffuse types of breast cancer such a lobular carcinomas, where most of the tumor mass consists of discohesive strands and scattered cells less reminiscent of epithelial cells.
E-cadherin expression is emerging as one of the pivotal caretakers of the epithelial phenotype.1 While E-cadherin expression is detectable in most differentiated breast cancers, there is an inverse relation with cancer grade and prognosis.13,14 E-cadherin is a transmembrane glycoprotein that mediates homophilic interactions between its extracellular immunoglobulin domains on neighboring cells, and connects indirectly to intracellular actin filaments via cytoplasmic catenins, forming a multiprotein complex with alpha- beta-, and p120 catenins (Fig. 2-2). Clusters of E-cadherin aggregate with each other, and other specialized proteins such as claudins, into junctional complexes to establish adherens junctions and desmosomes.1 In this way, E-cadherin is critical in establishing physical links between cells, and interconnecting them with their cytoskeleton and the basement membranes, establishing cell polarity and maintaining structural integrity and cellular signaling.
Figure 2-2
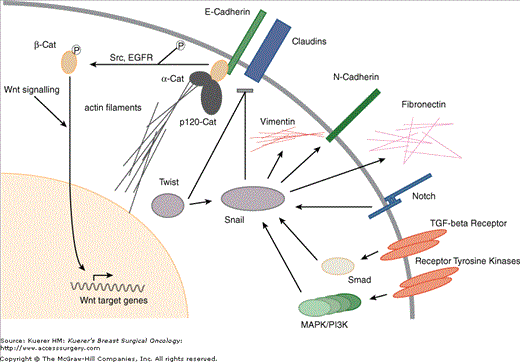
Simplified scheme of key regulators of epithelial-to-mesenchymal transition. The down-regulation of E-cadherin releases beta-catenins from their complexed state at the cell membrane, freeing them to participate in the Wnt signaling pathway, which cross-talks with several signaling cascades, including EGF/FGF, TGF-beta, and the hedgehog pathway. Snail has been placed at the center of a hub of several signaling pathways leading to EMT18. Snail is induced by numerous factors, including receptor tyrosine kinases, Notch signaling, and TGF-beta signaling, involving the PI3kinase, MAPkinase, and Smad pathways. Like Twist, another helix-loop-helix protein, it acts as direct inhibitor of E-cadherin transcription.
The key event promoting EMT is the down-regulation of E-cadherin, leading to a reversal of the effects listed above. In addition, decreasing levels of E-cadherin release beta-catenins from their complexed state at the cell membrane, freeing them to participate in the Wnt signaling pathway,15 which cross talks with the hedgehog signaling cascade, including EGF/FGF, TGF-beta, and Snail.2,3,16 The down-regulation of E-cadherin occurs during cancer progression through reversible and irreversible mechanisms, although mutations appear limited to lobular breast cancer17 and other diffuse neoplasms such as diffuse gastric cancer,2 where inactivating mutations are found in up to 50% of cases. More commonly, E-cadherin expression is down-regulated by promoter methylation-induced silencing, which may be dynamically reversible, as needed in the changing environment of cancer progression.5 Loss of E-cadherin can be accompanied by an increase in N-cadherin expression, known as cadherin switch. N-cadherin is typically expressed in mesenchymal cells, and enhances tumor motility through its interaction with the FGF receptor.17
While the control of epigenetic regulation, such as promoter hypermethylation, is incompletely understood, there is an inverse relation between transcriptional activity and the likelihood of epigenetic silencing. Direct transcriptional repression by transcription factors is a likely starting point, and several signaling pathways have been implicated, including members of the basic helix-loop-helix family such as E47 and Twist; and zinc finger transcription factors such as the Snail superfamily18 and ZEB1 and SIP1.2 One regulatory model suggests involvement of a different set of factors for the initiation of EMT, lead by Snail and SIP1, whereas Slug and E47 would maintain the EMT phenotype.19 Twist appears to have a critical role in enabling entry into the vasculature and favoring metastasis.20
Snail has been placed at the center of a hub of several signaling pathways leading to EMT, and may play a central role in breast cancer recurrence.18 Snail is directly induced by both receptor tyrosine kinase and TGF-beta signaling, involving the PI3kinase, MAPkinase, and Smad pathways.21 In addition to E-cadherin repression, Snail decreases desmoplakin, Muc-1, and cytokeratin-18 expression, while enhancing fibronectin and vimentin expression, all changes associated with EMT. Twist has been shown to induce Snail expression in some models,22 although in human tissues the evidence remains correlative at this time.23-25 Finally, Snail may play a central role in peritumoral stromal tissue as well, since serial analysis of gene expression (SAGE) showed it was one of the most abundantly expressed genes in tumor endothelial cells.26
Two additional transcription factors induced by TGF-beta signaling have recently been described. Goosecoid (GSC), a transcription factor that plays a central role in the Spemann neuroectodermal organizer,27 down-regulates E-cadherin and promotes metastasis in breast cells.28 Similar effects are observed with FOXC2, with induction of mesenchymal markers such as vimentin, fibronectin, and smooth muscle actin.29 Another homeodomain protein, HOXB7, which shows overexpression in breast cancer that increases with tumor progression to metastatic disease, has also been shown to induce EMT in breast cancer cell lines.30,31 Overexpression of HOXB7 induces several angiogenic and growth factors seen in EMT, such as FGF, VEGF, and MMP9. Table 2-1 lists some transcription factors modulating EMT in breast epithelial cells.
| Transcription Factor | DNA-Binding Domain | Reference | |
|---|---|---|---|
| E47 | Transcription factor 3 | Helix loop helix | 65 |
| FOXC2 | Mesenchyme forkhead 1 | Forkhead domain | 29 |
| GSC | Goosecoid | Homeobox | 28 |
| HOXB7 | Homeobox B7 | Homeobox | 31 |
| SIP1 | Smad interacting protein 1 | Zinc finger motif | 66 |
| SNAI1 | Snail | Zinc finger motif | 67 |
| SNAI2 | Slug | Zinc finger motif | 68 |
| Twist1 | Twist homolog 1 | Helix loop helix | 25 |
There are 3 general mechanisms responsible for the breach of the basement membrane separating the epithelial compartment from the surrounding stroma:
The increasing mechanical pressure arising from unrelenting proliferation
The increased motility of tumor cells undergoing EMT
The release of proteolytic enzymes breaking down the barrier
The extracellular matrix (ECM) is a product of stromal cells, and supports the adhesion and migration of cells through cell surface adhesion receptors, and provides support for the vasculature. The basement membrane is a specialized version of the ECM, separating both epithelia as well as blood vessels from the surrounding stroma. This provides a barrier to invasion by epithelial cells, and its penetration and destruction are prere-quisites for invasive tumor growth. The basement membrane is 20 to 200 nm thick, consisting of self-organizing networks of laminins and type IV collagen. A variety of additional glycoproteins and proteoglycans, including CD44, are present.
In order to break through this barrier, invading cancer cells secrete a wide variety of ECM-degrading proteases. The largest group consists of the matrix metalloproteinases (MMPs), of which 28 have been described so far. They each have their own specific degradation target proteins, such as various collagen subtypes, gelatins, fibronectin, elastin, laminin, and the critical basement membrane collagens. They have a role in regulating the tumor microenvironment, given that many of the breakdown products they generate have effects of their own in attracting epithelial and stromal cell migration.
Unlike oncogenes, MMPs are not up-regulated by amplifications or activating mutations but by transcriptional activation by factors such as Snail, SIP1, interferons, and growth factors. Conversely, expression of MMPs can be inhibited by differentiation factors and tumor suppressors, such as retinoids, corticosteroids, or TGF-beta.32 The expression and activation of MMPs is increased in most cancer types, including breast cancer.
A second major group of cancer invasion–associated proteases are the fibrinolytic proteinases, such as plasmin, plasminogen activators, and thrombin. These are produced remotely, such as in the liver or vasculature, and are deposited in tumors because of the hyperpermeability caused by changes in the ECM and basement membranes of tumor-associated blood vessels. They cleave fibrin and plasminogen, as well as laminin and type IV collagen. Other proteases include cathepsins, kallikreins, and elastase.
All of these proteases have counterbalancing antagonists, such as the class of tissue inhibitors of metalloproteases (TIMPs), and the serpin superfamily, which includes maspin and alpha-1 antitrypsin.33 In analogy to the balance between oncogenes and tumor suppressor genes in the control of the cell cycle, the effect of genes mediating invasiveness and metastatic spread is balanced by these inhibitory gene products, many of which promote cell anchorage and inhibit migration. It appears that the expression of these adhesion molecules, which include cadherins, is functionally dominant over the expression of receptors promoting invasion, and they are predominantly functionally silenced, rather than mutated, in invasive cancer.33
As invading tumor cells penetrate the basement membrane to gain access to the periductal stroma and the ECM, they enter a fundamentally new microenvironment. They are now directly exposed to stromal influence, and can engage not only in interactions with the extracellular matrix but also extensive reciprocal cross talk with adjacent fibroblasts, immune cells, and cells lining blood and lymphatic vessels.
Fibroblasts are primarily responsible for the synthesis and remodeling of the ECM, as well as for many soluble paracrine growth factors responsible for cell proliferation and neoplastic programming of tissues.34 Fibroblasts in peritumoral stroma are unique, and undergo dynamic changes during tumor progression.35 They are commonly referred to as cancer-associated fibroblasts (CAFs) or myofibroblasts, exhibit a relatively high proliferative index, express smooth muscle actin, and are often surrounded by dense accumulations of fibrillar collagens in a pattern referred to as desmoplasia.36 CAFs isolated from malignant tissue exhibit an altered phenotype characterized by enhanced production of collagens and epithelial growth factors.36
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


