Intracellular Pathways of Iodothyronine Metabolism/Implications of Deiodination for Thyroid Hormone Action
Antonio C. Bianco
Brian W. Kim
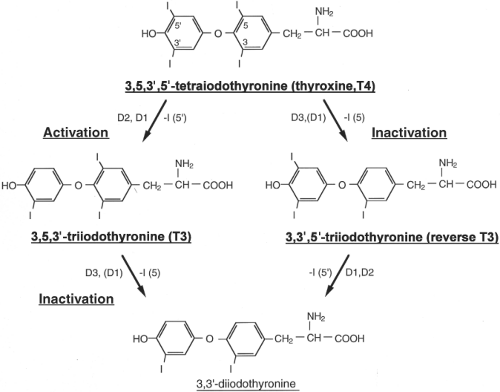 Figure 7.1 Structures and interrelationships between the principal iodothyronines activated or inactivated by the selenodeiodinases. |
Thyroxine (T4), the main product of the thyroid gland, is a prohormone that must be activated by deiodination of its phenolic outer ring, a conversion yielding triiodothyronine (T3). The latter form is bound by the thyroid hormone nuclear receptors with high affinity. The activating deiodination reaction is catalyzed by two deiodinases, type 1 (D1) and type 2 (D2) (Fig. 7.1). In humans, approximately 80% of the T3 that is produced each day is the result of extrathyroidal deiodination of T4, demonstrating the critical importance of these deiodinases for thyroid hormone homeostasis. In healthy subjects, the D2 pathway is thought to contribute more to plasma T3 than does D1; consequently, serum T3 concentrations decline only
modestly following treatment with propylthiouracil (PTU), a specific inhibitor of D1 (1). As a counterpoint to the activation pathway, both T4 and T3 can be converted to inactive forms via deiodination of their inner tyrosyl ring, a reaction catalyzed by either D1 or type 3 deiodinase (D3). About 40% of the T4, and nearly all of the T3 that is produced each day is eventually deiodinated by inner ring deiodination, mostly via D3 (Fig. 7.1).
modestly following treatment with propylthiouracil (PTU), a specific inhibitor of D1 (1). As a counterpoint to the activation pathway, both T4 and T3 can be converted to inactive forms via deiodination of their inner tyrosyl ring, a reaction catalyzed by either D1 or type 3 deiodinase (D3). About 40% of the T4, and nearly all of the T3 that is produced each day is eventually deiodinated by inner ring deiodination, mostly via D3 (Fig. 7.1).
Early work in the deiodinases field focused on their homeostatic role with respect to plasma T3 concentration. Iodine availability is likely to have been a major factor in the evolutionary pressure leading to the selection of the deiodinases for this purpose. The active form T3 is short-lived (half-life ∼1 day in humans); if T3 were the main product of thyroid secretion, its production would fluctuate according to the availability of iodine. Instead, the human thyroid mostly secretes T4, a long-lived molecule (half-life ∼7 days) that binds to circulating proteins and accumulates in a large extrathyroidal pool, approximately 770 μg (1 μmol) in adults. By allowing for dynamic conversion of T4 to T3, the deiodinase pathways decrease the sensitivity of thyroid hormone signaling to variations in iodine availability, adjusting in response to fluctuations in serum T4 concentrations such that constant serum T3 concentrations are maintained (2). That D1 can both activate and inactivate thyroid hormone has led to the speculation that it also plays a homeostatic role in maintaining iodine stores.
While the traditional homeostatic view focuses on the effects of deiodination on extracellular or plasma T3, the modern paradigm incorporates multiple lines of evidence indicating that deiodinases alter intracellular thyroid hormone signaling on a cell and tissue-specific basis (3). Rather than simply reacting to changes in plasma thyroid hormone levels, deiodinase activities can be directly induced or suppressed by a wide range of signaling pathways and pharmaceutical agents in human development, health, and disease states. The consequences of primary changes in deiodinase activities can be understood by following the fate of T3 and T4 in the plasma as they approach the target tissues: Cellular membranes are relatively impermeable to thyroid hormone and thus membrane transporters are necessary for access to the intracellular environment (4). Once inside the cells, thyroid hormone diffuses toward the nucleus and eventually binds to specific nuclear receptors, high affinity ligand-dependent transcription factors that modify gene expression (5). En route to the nucleus, however, T4 can be transformed to the biologically active T3 molecule by D2, thus increasing nuclear T3 supply without immediately affecting plasma T3 levels. Alternatively, if D3 were present, the T4 could be inactivated to form reverse T3, thus creating local hypothyroidism. Thus, the role of deiodinases in thyroid hormone action is both homeostatic with regard to plasma T3, but also primary and dynamic with regards to local, tissue-specific control of thyroid hormone signaling.
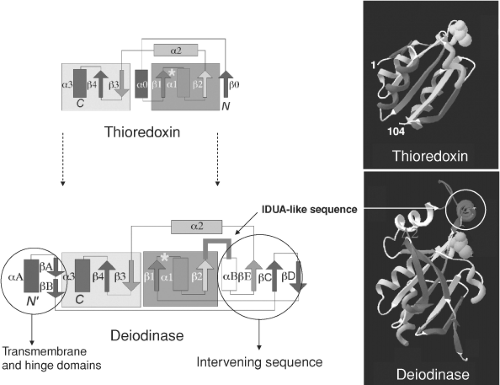 Figure 7.2 Three-dimensional structures and organization of secondary structure of the archetype thioredoxin enzyme and the rough model of the deiodinases deduced from the alignment revealed by hydrophobic cluster analysis. The transmembrane, hinge, and intervening sequences are circled. (Modified from Callebaut I, Curcio-Morelli C, Mornon JP, et al. The iodothyronine selenodeiodinases are thioredoxin-fold family proteins containing a glycoside hydrolase clan GH-A-like structure. J Biol Chem 2003;278:36887, with permission.) |
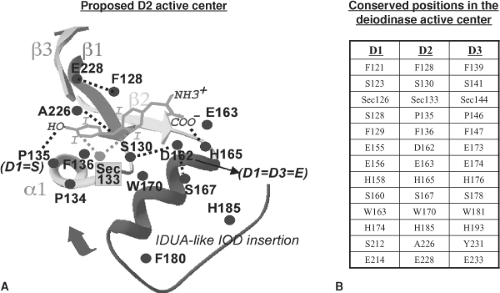 Figure 7.3 Schematic representation of the putative active site of deiodinases deduced from sequence alignment and from the associated modeling (see also Fig. 7.2). A: The positions shown are the conserved residues in the type 2 deiodinase (D2). B: Table contains the corresponding positions and residues in the other deidinases (D1 and D3). The iduronidase (IDUA)-like insertion likely constitutes a cap that may cover the active site after ligand binding. (Modified from Callebaut I, Curcio-Morelli C, Mornon JP, et al. The iodothyronine selenodeiodinases are thioredoxin-fold family proteins containing a glycoside hydrolase clan GH-A-like structure. J Biol Chem 2003;278:36887, with permission.) |
Structure of the Deiodinases
The three deiodinase proteins (D1, D2, and D3) are structurally similar (∼50% sequence identity), all being integral membrane proteins of 29 to 33 Kd (2,3,4). Because of inherent difficulties
in the crystallization of membrane-anchored proteins, further insight into the structures of these proteins has instead been obtained through the use of hydrophobic cluster analysis (6), a technique based on protein folding and two-dimensional transposition of sequences, allowing resolution of a sequence into its secondary structures centered on the so-defined hydrophobic clusters.
in the crystallization of membrane-anchored proteins, further insight into the structures of these proteins has instead been obtained through the use of hydrophobic cluster analysis (6), a technique based on protein folding and two-dimensional transposition of sequences, allowing resolution of a sequence into its secondary structures centered on the so-defined hydrophobic clusters.
On the basis of this type of analysis, the three deiodinases have a single transmembrane segment which is present near the N-terminus, and several well-conserved clusters, typical of α-helixes or β-strands, suggesting that they correspond to core secondary structures of the deiodinases. A striking common feature of the deiodinases is a shared resemblance with various members of the thioredoxin (TRX) family, proteins whose three-dimensional structure contains a fold defined by βαβ and ββα motifs (Fig. 7.2). As with other members of the thioredoxin family, the deiodinases contain extra elements not found in the canonical thioredoxin fold that interrupt the relationship between the βαβ and ββα motifs (7). A unique aspect of the deiodinases is that one of these intervening sequences has striking similarity to α-L-iduronidase (IDUA; 47% identity with D1 and D3 and 60% with D2), a lysosomal enzyme that cleaves α-iduronic acid residues from the glycosaminoglycans, heparan sulfate and dermatan sulfate (8) (Fig. 7.2). This structural similarity between the deiodinases and iduronidase may reflect the similarity of their substrates, T4 (or T3) and sulfated α-L-iduronic acid, respectively.
Three-dimensional modeling predicts that the active center of the deiodinases is a pocket defined by the ββ1-α1-β2 motifs of the thioredoxin fold and the IDUA-like insertion (Fig. 7.3).
The most striking feature of this pocket is the presence of the rare amino acid selenocysteine, which is critical for the catalytic activity of the three deiodinases. The presence of selenocysteine was first identified when rodent D1 complementary DNA (cDNA) was found to contain a UGA codon that encoded selenocysteine; in the vast majority of messenger RNAs (mRNAs), UGA is a stop codon (9). The cis-acting sequences that allow for the incorporation of selenocysteine rather than a stop codon consist of the selenocysteine codon (UGA) itself and a specific RNA stem–loop located in the 3′-untranslated region of the mRNA. Only in the presence of the stem–loop structure and in several trans-acting factors are UGA codons “recoded” to specify selenocysteine. The stem–loop sequence is termed selenocysteine (sec) insertion sequence, or SECIS element; it is present in all three deiodinases and all other eukaryotic selenoproteins (10). It would be expected that mutations in the genes controlling selenocysteine incorporation would have consequences for the deiodinases, and indeed families with mutations in selenocysteine insertion sequence-binding protein 2 (SBP-2) have abnormal thyroid test profiles (11,12).
The most striking feature of this pocket is the presence of the rare amino acid selenocysteine, which is critical for the catalytic activity of the three deiodinases. The presence of selenocysteine was first identified when rodent D1 complementary DNA (cDNA) was found to contain a UGA codon that encoded selenocysteine; in the vast majority of messenger RNAs (mRNAs), UGA is a stop codon (9). The cis-acting sequences that allow for the incorporation of selenocysteine rather than a stop codon consist of the selenocysteine codon (UGA) itself and a specific RNA stem–loop located in the 3′-untranslated region of the mRNA. Only in the presence of the stem–loop structure and in several trans-acting factors are UGA codons “recoded” to specify selenocysteine. The stem–loop sequence is termed selenocysteine (sec) insertion sequence, or SECIS element; it is present in all three deiodinases and all other eukaryotic selenoproteins (10). It would be expected that mutations in the genes controlling selenocysteine incorporation would have consequences for the deiodinases, and indeed families with mutations in selenocysteine insertion sequence-binding protein 2 (SBP-2) have abnormal thyroid test profiles (11,12).
All three deiodinases seem to function as homodimers (13). In early studies using gel filtration and gradient centrifugation of solubilized kidney or liver microsomal membranes, deiodinase activity was detected in higher molecular weight forms than predicted from their respective deduced amino acid sequences (29 to 33 kDa) (14,15). Co-expression of an inactive rat D1 Sec126Ser protein in porcine LLC-PK1 cells with endogenous (active) D1, followed by the immunoprecipitation of D1 activity using a rat D1-specific antibody indicated that D1 homodimerization occurs in cells (16). Subsequent studies using three independent approaches in a cellular model transiently expressing the deiodinases demonstrated the existence of D1, D2, and D3 dimers (17). Deletion/truncation analysis coupled with immunodepletion assays suggested that a conserved C-terminal region of D1 corresponding to rat amino acids 148 to 163 serves as a dimerization domain responsible for posttranslational assembly of deiodinases (18). Further characterization of this domain using alanine scanning mutagenesis identified 152IYI154 as critical residues required for D1 dimer assembly (19). Use of a similar strategy identified five residues (153FLIVY157) at the beginning and three residues (164SDG166) at the end of a homologous region of D2 as required for dimerization (19).
Among the deiodinases, dimerization is the best understood for D2. Live-cell fluorescence resonance energy transfer (FRET) and bioluminescence resonance energy transfer were used to study the interaction between D2 domains in the D2:D2 homodimer (20). D2 has a typical transmembrane helix containing potentially charged residues (D, E, K, R, H) that can be stabilized in the hydrophobic membrane environment by dimerization. In this way, charged residues could be neutralized via an intermolecular interaction of residue couples such as D29/K35 as well as direct contacts between polar residues such as H36-K35′, which is compatible with modeling of the two D2 transmembrane segments in an αα dimeric architecture. In support of this, a truncated D2 molecule missing the first 42 amino acids is a cytosolic protein that does not homodimerize. In addition to this critical role played by the transmembrane domain, interaction at the globular domain has also been demonstrated because a full-length D2 molecule dimerizes with the truncated D2 monomer lacking the transmembrane domain (20).
Further characterization of the deiodinase globular dimerization interface was performed by taking advantage of the finding that deiodinases are TRX fold-containing proteins (6). The deiodinase globular interacting interface was tentatively modeled by comparison to the human TRX fold (20) and shows a clear propensity to dimerize via a large area formed by the alignment of two β strands that constitute a small β sheet (21). Due to the high sequence identity around the canonical TRX β1-α1-β2 motif, the deiodinase globular dimerization model could be fitted on the crystal structure of dimeric oxidized TRX with no evident conflict. These studies indicate that the native deiodinase dimer is formed by interactions at both the transmembrane and the globular cytosolic domains. The fact that dimerization is critical for catalytic activity indicates that proper conformation for each active center can only be achieved in the dimeric state.
Topology and Subcellular Localization
The D1 and D2 monomers are integral membrane proteins oriented with a small amino-terminal extension in the extracellular space (D1) or lumen of the endoplasmic reticulum (D2), and a single transmembrane domain exiting the membrane at about position 40 (22,23). This puts the active center of both D1 and D2 in the cytosol. D3 is also an integral membrane protein. Although initial findings indicated that its active center is in the extracellular space (24), more recent analysis indicates that D3’s topology is similar to that of the other deiodinases, with the active center in the cytosol (Fig. 7.4). This is also supported by the observation that D3-mediated T3 deiodination is much more efficient when cells are co-expressing MCT8, the transporter that grants T3 access to the intracellular compartment (25).
The location of mature D1 in the plasma membrane has been demonstrated in several types of cells that normally contain D1, including thyroid and kidney cells, and in cells transiently expressing the enzyme (16,26,27,28). Using confocal laser microscopy of human and mouse cells, transiently expressed FLAG-tagged D1 was also localized to the plasma membrane. D1 does not colocalize with the endoplasmic reticulum protein BiP, as does D2 (23). This plasma membrane localization has been confirmed by labeling of D1 with cell-impermeable reagents (24). D2, on the other hand, is an endoplasmic reticulum protein. Immunofluorescent confocal microscopy of human and mouse cells transiently expressing FLAG-tagged D2 revealed colocalization of D2 with BiP. Endogenously expressed D2 also colocalizes with BiP in MSTO-211H cells (29).
Using similar techniques, endogenously and transiently expressed FLAG-tagged D3 was identified in the plasma membrane. It colocalizes with the α-subunit of Na+, K+-ATPase, the early endosomal marker EEA-1, and clathrin, but not with endoplasmic reticulum proteins. There is constant internalization of D3 that is blocked by sucrose-containing medium, and exposing cells to a weak base such as primaquine increases the pool of internalized D3, suggesting that D3 is recycled between plasma membrane and early endosomes. Such recycling could account for the longer half-life of D3 (12 hours), as compared with D1 (8 hours) or D2 (1 hour) (24).
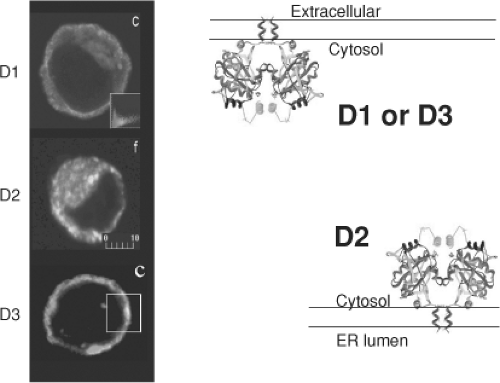 Figure 7.4 Subcellular localization and topology of D1, D2, and D3. Left panel: Immunofluorescence confocal microscopy of HEK-293 cells transiently expressing one of the deiodinases as indicated. D1 is visible in the periphery of the cell, and does not colocalize with BiP, an endoplasmic reticulum marker visible around the nucleus. D2 is visible colocalized with BiP, whereas D3 is visible in the cell border. Right panel: Schematic representation of type 1 deiodinase (D1), D2, and D3 dimers with their respective orientations and subcellular localization. (From Toyoda N, Berry MJ, Harney JW, et al. Topological analysis of the integral membrane protein, type 1 iodothyronine deiodinase (D1). J Biol Chem 1995;270:12310; Baqui MM, Gereben B, Harney JW, et al. Distinct subcellular localization of transiently expressed types 1 and 2 iodothyronine deiodinases as determined by immunofluorescence confocal microscopy. Endocrinology 2000;141:4309>; and Baqui MM, Botero D, Gereben B, et al. Human type 3 iodothyronine selenodeiodinase is located in the plasma membrane and undergoes rapid internalization to endosomes. J Biol Chem 2003;278:1206, with permission.) |
In contrast to D1, D2 in located in the endoplasmic reticulum in the same cell types (23). This differential subcellular localization of D1 and D2 may explain the small contribution of T3 generated by D1 and the large contribution of T3 generated by D2 to intranuclear T3 (30,31,32). The plasma membrane location of D3 makes it readily accessible to the T4 and T3 entering the cells, explaining its capacity for rapid dampening of thyroid hormone signaling in D3-expressing cells (33). The plasma membrane location could also explain the efficiency with which D3 overexpression in patients with hemangiomas inactivates thyroid hormone as well as its blockade of the access of maternal thyroid hormones to the fetus (34,35).
Structure–Function Relationships of Deiodinases and Mechanism of Deiodination
The deiodination of iodothyronines by D1, D2, and D3 is a reductive dehalogenation that requires a reducing agent, such as dithiothreitol in vitro, as a cofactor. An endogenous cofactor capable of sustaining multiple rounds of iodothyronine deiodination has not yet been identified. On the basis of major differences in substrate preference, reaction kinetics, and sensitivity to various inhibitors, it was assumed that the substrate-binding pockets of these enzymes were fundamentally different. However, the structural model described above predicts a conserved structure for all three deiodinases, particularly for the active center, the accuracy of which is supported by the observation that substantial perturbations in enzyme function result from changes of single amino acids in the binding pocket (6) (Fig. 7.3).
D1 has a relatively low affinity for T4 [Michaelis–Menten constant (Km), 1 to 2 μM], and its catalytic activity is bisubstrate in nature, with a thiol-containing cofactor serving as the second substrate with “ping-pong” kinetics (36) (Fig. 7.5). D2 and D3, on the other hand, have relatively high affinity for T4 and T3 respectively (Km, 1 to 4 nM), and both have sequential reaction kinetics, suggesting that the iodothyronine and the thiol-containing cofactor interact with the enzyme simultaneously before the reaction takes place (36). Regardless of the reaction kinetics, the selenocysteine residue in the active center of the three deiodinases probably acts as a nucleophile, catalyzing the removal of iodine. The critical role of selenocysteine in this function was ascertained through characterization of the kinetic properties of the wild-type selenoenzymes (D1, D2, and D3) and the corresponding cysteine (Cys) mutants, which have lower affinity for the substrates and decreased turnover rates (17,37,38).
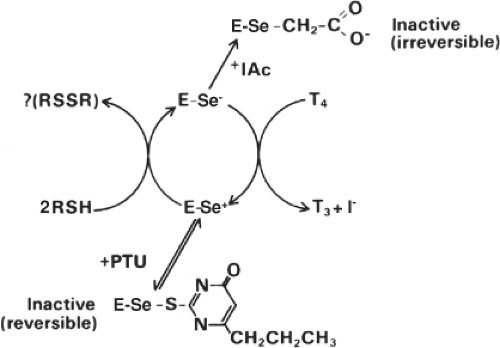 Figure 7.5 Mechanism of type 1 deiodinase (D1)-catalyzed thyroxine (T4) conversion to triiodothyronine (T3). The steps in the enzymatic reaction cycle at which iodoacetic acid (IAc) and propylthiouracil (PTU) are thought to inhibit catalysis are indicated. (From Leonard JL, Rosenberg IN. Thyroxine 5′-deiodinase activity of rat kidney: observations on activation by thiols and inhibition by propylthiouracil. Endocrinology 1978;103:2137, with permission.) |
D1, D2, and D3 differ in their sensitivity to PTU. D1 is quite sensitive [inhibition constant (Ki), 5 μM; in vitro at 10 μM dithiothreitol], but D2 and D3 are not (Ki >1 mM). PTU probably inhibits D1 by competing with the endogenous thiol-containing cofactor for a putative selenenyl iodide (E-Se-I) intermediate (36) (Fig. 7.5). Supporting this interpretation is the fact that PTU inhibition is uncompetitive with the first D1 substrate (iodothyronine), but competitive with the second (e.g. dithiothreitol) (36). Because of the PTU insensitivity, D2- or D3-catalyzed deiodination proceeds by removal of an iodonium (I+) ion by the endogenous cofactor, resulting in an enzyme–thyronine intermediate [D2-T3 (or reverse T3)] complex and a cofactor-Se-I complex (39). In this regard, it is notable that the sequence Thr-Sec-Pro-Pro/Ser-Phe is identical in D1, D2, and D3, but that in all D1 sequences, excepting
the PTU-insensitive D1 of the blue tilapia (Oreochromis aureus), the uncharged polar side chain of the Ser residue substitutes for the nonpolar side chain of Pro at position 128 (40). The existence of this natural variation prompted the creation of the Ser128Pro D1, Pro135Ser D2, and Pro146Ser D3 proteins (Fig. 7.3). Remarkably, replacement of Pro135 with Ser in D2 results in two orders of magnitude increase in Km (T4) to approximately 250 nM, approximately tenfold lower than that of D1 for T4 and the enzyme operates with ping-pong kinetics. Furthermore, the Pro135Ser D2-catalyzed deiodination is two orders of magnitude more sensitive to PTU (Ki 4.0 μM), although PTU inhibition is noncompetitive with dithiothreitol. Thus, the substitution of Ser for Pro 135 in D2 results in changes in the enzyme that make its kinetics more similar to those of D1, indicating a critical influence of the amino acid occupying this position on enzyme function. Similar observations were made with the Pro146Ser D3 protein, which has a fivefold higher Km (T3) and is highly sensitive to inhibition by PTU (Ki 1.0 μM). As mentioned above, Ser128Pro D1 catalyzed deiodination is resistant to PTU (Ki >1 mM), suggesting that there is no longer an accessible E-Se-I intermediate, again illustrating the pivotal role of this position in the active center of the deiodinase molecule.
the PTU-insensitive D1 of the blue tilapia (Oreochromis aureus), the uncharged polar side chain of the Ser residue substitutes for the nonpolar side chain of Pro at position 128 (40). The existence of this natural variation prompted the creation of the Ser128Pro D1, Pro135Ser D2, and Pro146Ser D3 proteins (Fig. 7.3). Remarkably, replacement of Pro135 with Ser in D2 results in two orders of magnitude increase in Km (T4) to approximately 250 nM, approximately tenfold lower than that of D1 for T4 and the enzyme operates with ping-pong kinetics. Furthermore, the Pro135Ser D2-catalyzed deiodination is two orders of magnitude more sensitive to PTU (Ki 4.0 μM), although PTU inhibition is noncompetitive with dithiothreitol. Thus, the substitution of Ser for Pro 135 in D2 results in changes in the enzyme that make its kinetics more similar to those of D1, indicating a critical influence of the amino acid occupying this position on enzyme function. Similar observations were made with the Pro146Ser D3 protein, which has a fivefold higher Km (T3) and is highly sensitive to inhibition by PTU (Ki 1.0 μM). As mentioned above, Ser128Pro D1 catalyzed deiodination is resistant to PTU (Ki >1 mM), suggesting that there is no longer an accessible E-Se-I intermediate, again illustrating the pivotal role of this position in the active center of the deiodinase molecule.
The presence of Pro in the 128/135/146 positions of D1, D2, and D3 may result in tighter binding of the substrate in the D2 (and D3) binding pockets, perhaps explaining the approximately 1000-fold higher affinity of D2 and D3 for T4 as compared with D1. This could reflect an interaction between the phenolic hydroxyl group of T4 and the Pro residue at these positions, and explain why sulfate conjugation of the phenolic hydroxyl group dramatically increases the maximum velocity (Vmax)/Km and changes the T4 deiodination site from an outer to an inner ring iodine (41).
Three conserved amino acids with charged polar side chains mark the transition between the β2-strand and the a-helix in the IDUA-like insertion, Glu/Asp155, Glu156, and His158 (D1 residues). When the invariant Glu156 (D1), Glu163 (D2), and Glu174 (D3) were replaced with Ala, the resulting enzymes had no deiodinase activity. Replacement of Glu156 with Asp in D1 supported deiodination, but with an approximately 4.5-fold higher Km (rT3), whereas a similar substitution at position 163 in D2 did not alter the Km (T4). Thus, the acidic amino acids in this region of the deiodinase pocket are important for substrate binding or enzyme function, although the length of the side chain can vary. This is further supported by mutational studies of His at position 158 in D1: Mutation to Asn, Gln, or Phe resulted in complete loss of deiodinase activity (42). Replacement of the corresponding His 165 in D2 with Asn also resulted in loss of deiodinase activity. According to the model, residues in this acidic pocket could interact with either the amino or the carboxyl group in the alanine side chain of the iodothyronines, a hypothesis supported by previous studies indicating that the positively charged T4 analogue (3,5,3′, 5′-tetraiodothyroethylamine), which lacks a carboxyl group, is not a substrate for D1 (43). In addition, the compounds with the highest affinity for D1 (lowest apparent Km values) are those that lack positively charged functional groups (NH3+), such as tetraiodothyroacetic acid. Furthermore, the Km (T4) values for D- and L-T4 are similar (43). These results argue that the carboxyl group in the iodothyronines interacts with the NH3 group of His in the 158 position of D1 and that the other acidic residues in this pocket act to reduce the ionization of the His residue. The critical role played by the IDUA-like insertion is further strengthened by the complete loss of deiodinase activity when the conserved Trp163 in D1 and the corresponding Trp 170 in D2 are replaced with Ala.
Specific Properties of the Deiodinases
Type 1 Deiodinase
PTU-inhibitable, D1-catalyzed conversion of T4 to T3 supplies approximately 20% of the T3 in the serum in normal humans, but 50% or more in patients with thyrotoxicosis. D1 is the only selenodeiodinase that can function as either an outer (5′)- or inner (5)-ring iodothyronine deiodinase, D2 and D3 being exclusively outer and inner ring deiodinases, respectively (44). The molecular basis for these differences is not known.
The gene (Dio1) for human D1 is on chromosome 1 p32-p33, in a region syntenic with mouse chromosome 4, the location of mouse Dio1 (45). The complete cDNA sequences have been determined for rat, human, mouse, dog, chicken, and tilapia D1 (9,40,46,47,48,49). The size of the mRNAs for these D1s is about 2 to 2.1 kb, and all contain a UGA codon in the region encoding the active center, which is highly conserved among species. By Northern analysis, D1 is expressed in many tissues of most vertebrates (50,51,52). In rats, these include the liver, kidneys, central nervous system (CNS), anterior pituitary gland, thyroid gland, intestine, and placenta. In humans, D1 activity is notably absent from the CNS, but is present in the liver, kidneys, thyroid, and pituitary (53,54). At the same time, clues obtained from the D1 knockout mouse hint that D1 might also affect thyroid economy by playing a scavenger role in iodine metabolism (55). Conjugated iodothyronines are the preferred substrates for D1 and have increased water solubility and are thus preferentially eliminated in the urine and bile. However, loss of iodine through these pathways is minimized because conjugated iodothyronines are excellent substrates for D1, which is highly expressed in liver and kidneys (55).
Regulation of D1 Synthesis
Changes in D1 activity have been investigated in developing rats; in general, D1 activity is low in all tissues of fetal rats. It appears soon after birth in the intestine, liver, kidneys, cerebrum, cerebellum, and gonads (56). The age-related changes in D1 mRNA content are similar, indicating that the changes in activity arise at a pretranslational level. The mechanism for the age-related changes in D1 expression is unknown. The physiologic benefit of the low D1 activity in the fetus is presumably low serum T3 concentrations, thus permitting changes in intracellular T3 to be determined by the developmentally programmed changes in D2 and D3 activity (57).
Thyroid Hormone
Thyroid hormone–induced increases in D1 activity and mRNA content are well documented in rats, mice, and humans (9,58). The increases are due to increased transcription, which in the human Dio1 gene can be attributed to the presence of two thyroid hormone–response elements (TREs) in the 5′-flanking region of the gene (59,60,61). Studies in thyroid receptor (TR) knockout mice indicate that the T3-induced D1 stimulation is largely mediated by the β subtype
of the receptor (62). Given this finding, the levels of the human Dio1 gene to T3 would be expected to be greatest in patients with thyrotoxicosis. In fact, the D1 mRNA content of peripheral blood mononuclear cells is increased in proportion to the degree of thyrotoxicosis (54). This increase can explain the acute decrease in serum T3 concentrations that occurs in response to PTU in patients with thyrotoxicosis (63).
of the receptor (62). Given this finding, the levels of the human Dio1 gene to T3 would be expected to be greatest in patients with thyrotoxicosis. In fact, the D1 mRNA content of peripheral blood mononuclear cells is increased in proportion to the degree of thyrotoxicosis (54). This increase can explain the acute decrease in serum T3 concentrations that occurs in response to PTU in patients with thyrotoxicosis (63).
Cytokines
Interleukin-1 (IL-1), IL-6, tumor necrosis factor-α (TNF-α), and other cytokines are potential mediators of the alterations in thyroid function that occur in patients with severe nonthyroidal illness (see section on nonthyroidal illness in Chapter 11C) (64,65,66). TNF-α, IL-1β, and interferon-γ decrease D1 activity and mRNA in rodent thyroid (FRTL5) cells (67). The effects of TNF-α have been examined in hepatocytes and HepG2 cells with contradictory results. TNF-α decreased the stimulatory effect of T3 on D1 mRNA production in HEPG2 cells, an action that is blocked by nuclear factor kappa B (68). In dispersed rat hepatocytes, IL-1β and IL-6 blocked T3 stimulation of D1 mRNA and activity; however, TNF-α had no effect (69). The effect of IL-1β was blocked by coexpression of the nuclear steroid receptor co-activator-1 (SRC-1), but not by cyclic AMP responsive element-binding protein (CBP) or CBP-associated factor (pCAF). Because IL-1 did not alter the amount of SRC-1 in hepatocytes, the effect was attributed to competition between IL-1 and T3-stimulated transcriptional events for limiting quantities of SRC-1. More recent studies describing decreases in D1 activity in liver and possibly D2 activity in skeletal muscle and increases in D3 activity in liver and skeletal muscle in patients dying of multiple organ failure and related conditions are discussed in the section on nonthyroidal illness in Chapter 11C.
Nutritional Influences on D1 Expression
A decrease in serum T3 concentrations relative to those of T4 and an increase in serum reverse T3 concentrations during fasting in humans was one of the earliest indications that the peripheral metabolism of thyroid hormones in humans was modulated by physiologic or pathophysiologic events (70). Similar changes occur in virtually all acutely ill and many chronically ill patients (71,72). Because thyroidal secretion accounts for only about 20% of daily T3 production in humans, the illness-associated decrease in serum T3 concentrations must be caused, largely if not completely, by decreased T4 to T3 conversion by D1 or D2 or by increased T3 clearance by D3 (73,74).
Early studies of liver D1 activity in rats suggested that the decrease in T4 conversion to T3 that occurred during fasting might be caused by a decrease in the thiol cofactor that serves as the cosubstrate for D1-catalyzed T4 to T3 conversion (75,76). However, this cofactor has not been identified. Although rats have been studied extensively as a model for the effects of fasting (and illness) on T4, to T3, conversion in humans, they are a poor model for the effects of fasting in humans because of their low body fat content and the fact that, unlike humans, their serum TSH and T4 concentrations decrease rapidly when they are starved (77). Also, despite reduced hepatic D1 activity, total body conversion of T4 to T3 is not reduced during starvation in rats (78,79). The marked fasting-induced reduction in serum TSH, T4, and T3 concentrations (i.e., central hypothyroidism) in rats is probably due, at least in part, to leptin deficiency (80). Prefeeding rats with a high-fat diet to induce obesity results in less urinary nitrogen loss and a lesser decline in serum T4 and T3 concentrations during starvation, and serum T3 concentrations actually increase if the period of starvation is prolonged (77). In contrast, in humans, serum T3 concentrations decrease rapidly to about 50% of baseline during fasting, and they remain low for up to 3 weeks of fasting, but serum T4 and TSH concentrations change little (81).
Selenium Availability
A decrease in hepatic D1 activity of Se-deficient rats and the demonstration that D1 could be labeled with 75Se were the first clues that this trace element was critical to the function of D1 (82,83,84,85). However, the effects of Se deficiency on the synthesis of intracellular selenoproteins, such as the selenodeiodinases, depend on the tissue being examined. For example, in Se-deficient rats, thyroidal D1 activity is preserved, while that in the liver declines precipitously (86), serum T3 concentrations increase, and serum T4 concentrations do not change (87). Se deficiency also decreases D1 activity in the kidneys; this is accompanied by a decrease in D1 mRNA, which does not occur in the liver (88). Se deficiency can occur in patients receiving diets that are restricted in protein content, such as those given for phenylketonuria, and has also been found in elderly patients (89,90,91,92). In Se-deficient humans, serum T4 concentrations and the serum ratio of T4 to T3 are slightly increased, but serum TSH concentrations are normal. In one endemic goiter region in Africa, there is an accompanying Se deficiency (93,94). When Se was supplied to these iodine-deficient people, their thyroid function deteriorated, as evidenced by an increase in serum TSH concentrations and a decrease in serum T3 concentrations, suggesting that the reduction in D1 activity during Se deficiency can protect against iodine deficiency, presumably by reducing inner ring deiodination of T4 or T3 (95,96).
H2 Type 2 Deiodinase
Type 2 deiodinase is an obligate outer ring selenodeiodinase that catalyzes the conversion of T4 to T3. The Km of D2 for T4 is in the nanomolar range under in vitro conditions in the presence of 20 mM dithiothreitol. The presence of D2 activity in many tissues, including bone, skin, and skeletal muscle provides a plausible source for a substantial fraction of the extrathyroidally generated T3 in human serum (38). T4 causes a posttranslational decrease in D2 activity because catalysis induces ubiquitin conjugation to D2, inhibiting the enzyme (97).
Gene Structure and Chromosomal Localization
The Dio2 gene is present as a single copy located on the long arm of chromosome 14 (14q24.3) in humans (98,99). It is about 15 kb in size, and the coding region is divided into two exons by an intron of approximately 7.4 kb. The exon/intron junction is located in codon 75, and is at the same position in the human and mouse Dio2 genes (98,100,101). For the human gene, there are three transcriptional start sites, 707, 31, and 24 nucleotides 5′ to the initiator codon ATG. The longest 5′-untranslated region of human D2 mRNA contains an intron of approximately 300 bp that can be alternatively spliced (101). Other splicing variants involving the coding region have also been identified (102). The human, mouse, and rat Dio2 5′-flanking regions have been isolated and functionally characterized. All contain a functional cyclic adenosine monophosphate
(cyclic AMP) response element, but only human Dio2 has binding sites for thyroid transcription factor-1 (TTF-1) (101,103,104).
(cyclic AMP) response element, but only human Dio2 has binding sites for thyroid transcription factor-1 (TTF-1) (101,103,104).
D2 mRNA and Protein
Human, mouse, and chicken D2 cDNAs containing intact 3′-untranslated regions (5-to 7.5-kb) have been identified using GenBank searches and library screening. These D2 cDNAs encode functional D2 proteins, as determined by expression in Xenopus laevis oocytes (100,105,106). Rat and human D2 mRNAs are approximately 7.5 kb, and chicken cDNA is approximately 6 kb (38,105,106,107). A detailed analysis involving nuclease mapping, primer extension, and Northern blots indicated that human D2 mRNA exists as four different transcripts in thyroid, brain, and possibly other tissues (101). The longest transcript is approximately 7.5 kb, starts 708 nucleotides upstream from the initiator ATG codon, and is the only transcript found in placenta. A shorter (approximately 7.2 kb) minor D2 species uses the same transcriptional start site, but the approximately 300-bp intron is spliced out. Two shorter transcripts of approximately 6.8 kb, differing by only seven nucleotides, use 3′ transcriptional start sites close to the translation initiation site. It is not known whether the rat and mouse genes use the same two major transcriptional start sites, but this is likely to be so for D2 in rat brain (38,101,107). The deduced amino acid sequences of the chicken, mouse, rat, and human D2 enzymes contain two selenocysteine residues. The first is in the active center of the enzyme, whereas the second is located close to the carboxy-terminus. In fish and frog D2, there is only one selenocysteine codon, which is located in the active center of the enzyme (38,100,106,107,108,109). Truncating the carboxy-terminal amino acids, including the carboxy-terminal selenocysteine, in human D2 has no effect on D2 enzyme kinetics or activity (110).
Tissue Distribution
In rodents, D2 activity is predominantly expressed in the pituitary, brain, and brown adipose tissue (BAT) (32,111,112,113,114,115). D2 activity is also present in the bone, gonads, pineal, thymus, and uterus of rats, mammary gland of mice, and vascular smooth muscle cells in humans (56,116,117,118,119,120,121). High levels of D2 mRNA and activity are found in the mouse cochlea at the eighth postnatal day, suggesting a role for D2 in generating T3 for cochlear development (122). In the cerebral cortex of neonatal rats, D2 mRNA is present in astrocytes (123) and tanycytes; the latter are specialized ependymal cells lining the third ventricle that have multiple cellular processes that express D2 mRNA and that extend to the median eminence (123,124,125,126). A monosynaptic pathway has also been identified between the arcuate nucleus, which contains D2, and the paraventricular nucleus, which contains thyrotropin-releasing hormone (TRH) (127).
In humans, D2 mRNA or activity is expressed not only in vascular smooth muscle cells, but also in the thyroid, heart, brain, spinal cord, skeletal muscle, and placenta, and small amounts of D2 mRNA have been detected in the kidneys and pancreas (38,101,107,128,129,130). Thyroid tissue contains relatively more D2 mRNA than D2 activity, with the exception of thyroid tissue in patients with thyrotoxicosis caused by Graves’ disease and follicular adenomas, in which both D2 mRNA and activity are present in large amounts (128). The discrepancy between D2 mRNA and activity is probably due to substrate-induced D2 ubiquitination. D2 mRNA sequences are also present in libraries from prostate, breast, and uterus, but none of these tissues have D2 activity (105). D2 mRNA or activity is present in human pituitary glands and brain tumors (128,131,132,133), and D2 activity has been found in human keratinocytes (134) and mesothelioma cells (29).
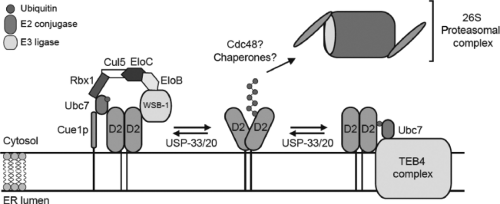 Figure 7.6 Representation of ubiquitination and proteasomal degradation of type 2 deiodinase (D2). Ub, ubiquitin; El, enzyme that activates Ub; E2, Ub conjugase; Ubc6 and Ubc7, E2s involved in D2 ubiquitination; Cue1, endoplasmic reticulum-docking protein for Ubc7; asterisk in the D2 molecule, selenocysteine-containing active center; isopeptidase catalyzes D2 deubiquitination. |
Regulation of D2 Synthesis
The Dio2 gene is regulated by a cyclic AMP-mediated pathway. Cold exposure increases D2 mRNA and activity in BAT in rodents, and α1-or β-adrenergic antagonist agents block this effect (115). In isolated brown adipocytes, the increase of D2 activity during catecholamine treatment is actinomycin D sensitive (135,136,137). In addition, D2 activity in BAT is induced by norepinephrine, isoproterenol, insulin, and glucagon, and it is inhibited by growth hormone (138,139). Cyclic AMP increases D2 activity in mesothelioma cells (29) and in rat astroglial cells (112,140), as does both nicotine and cyclic guanosine monophosphate (cyclic GMP) (141,142). As noted above, D2 mRNA and activity are increased in thyroid tissue from patients with Graves’ thyrotoxicosis, and forskolin increases D2 mRNA in dispersed human thyroid cells (101,128). It is therefore not surprising that human, rat, and mouse Dio2 contains a cyclic AMP response element approximately 90 nucleotides upstream of the transcriptional start site (101,103,104,143). The promoter activity of human Dio2 increases tenfold when cells are co-transfected with Dio2 and the α-catalytic subunit of protein kinase A. Mutation of the latter element abolishes the effect and decreases basal expression of Dio2 by approximately 90% (101).
Acting via a plasma membrane receptor known as TGR5, bile acids can also stimulate the cAMP pathway and activate D2 expression in BAT and skeletal muscle (144). This has been shown to activate thermogenesis and protect against diet-induced obesity. In addition, certain flavonols such as kaempferol also cause cAMP accumulation and D2 induction in BAT and skeletal myocytes (145), opening the interesting possibility that pharmacological intervention of this pathway might have clinical benefits in the treatment of obesity.
Although there is a high level of D2 mRNA in human thyroid tissue, no D2 mRNA or activity is present in FRTL-5 rat thyroid cells, and in adult rat thyroid tissue, D2 mRNA levels are very low and D2 activity is undetectable (104,128,146). Expression of the Dio2 gene in human thyroid tissue is under the control of TTF-1 but is not affected by Pax-8 (104). The human Dio2 gene has two TTF-1 binding sites, which are not present in the rat Dio2 gene, despite an overall 73% cross-species homology. The lack of these sites may explain the very low expression of D2 mRNA and activity in rat thyroid tissue.
Regulation of Degradation of D2
D2 is the critical T3-generating deiodinase due to its substantial responsiveness to physiologic signals. For example, D2 responsiveness to cyclic AMP constitutes the basis for the adrenergic stimulation of D2 activity in BAT, and human skeletal muscle and thyroid tissue. This links D2 expression with the sympathetic nervous system and widens the spectrum of environmental and endogenous stimuli that can potentially influence adaptive T3 production (see reference 1 for review).
Several transcriptional and posttranslational mechanisms have evolved to ensure tight control of tissue levels of D2, which
is inherent to its homeostatic function. The D2 mRNA in higher vertebrates is more than 6 kb in length, containing long 5′ and 3′ untranslated regions. The D25′ untranslated regions are greater than 600 nucleotides in length, and they contain three to five short open reading frames, which reduce D2 expression by as much as fivefold (147). Alternative splicing is another mechanism that regulates the level of D2 synthesis, because mRNA transcripts similar in size to the major 6- to 7-kb D2 mRNAs, but not encoding an active enzyme, are present in both human and chicken tissues (147).
is inherent to its homeostatic function. The D2 mRNA in higher vertebrates is more than 6 kb in length, containing long 5′ and 3′ untranslated regions. The D25′ untranslated regions are greater than 600 nucleotides in length, and they contain three to five short open reading frames, which reduce D2 expression by as much as fivefold (147). Alternative splicing is another mechanism that regulates the level of D2 synthesis, because mRNA transcripts similar in size to the major 6- to 7-kb D2 mRNAs, but not encoding an active enzyme, are present in both human and chicken tissues (147).
The ratios of D2 activity to D2 mRNA level in tissues vary, indicating substantial posttranslational regulation of D2 expression (148). In fact, the decisive property of D2 that characterizes its homeostatic behavior is a half-life of approximately 40 minutes that can be further reduced to approximately 25 minutes by exposure to physiologic concentrations of its substrate, T4, or extended to approximately 300 minutes when cells are grown in medium lacking T4 (149,150,151,152,153,154,155,156). This constitutes a rapid, potent regulatory feedback loop that efficiently controls T3 production and intracellular T3 concentrations based on how much T4 is available. The potency of the T4 in inducing loss of D2 activity mirrors the enzyme’s affinity for the substrate, indicating that enzyme–substrate interaction must occur to induce loss of D2 activity.
At the molecular level, D2 activity is regulated by conjugation to ubiquitin, a protein of approximately 8 Kd. The ubiquitinated D2 is subsequently recognized and degraded by proteasomes (157,158) (Fig. 7.6). The first evidence for this process was obtained in GH4C1 cells, in which the half-life of endogenous D2 was noted to be stabilized by MG132, a proteasome inhibitor (159). Substrate-induced loss of D2 activity was also inhibited by MG132 in these cells, indicating that both pathways affecting loss of D2 activity were mediated by the proteasomes. This implies that the loss of D2 activity is at least partially due to proteolysis of D2, a premise that was confirmed when the levels of immunoprecipitable D2 were found to parallel D2 activity both under basal conditions and after exposure to T4 (160). In subsequent studies it became clear that D2 is ubiquitinated (97), and the various enzymes involved in this process were identified. In studies in which human D2 was expressed in yeast, Ubc6p and Ubc7p were identified as the ubiquitin conjugases involved in ubiquitination of D2 (161), and it is now clear that these conjugases play a role in ubiquitination of human D2 (162,163).
Fusion of the 8-amino acid FLAG sequence to the carboxyl-, but not the amino-, terminus of D2 prolongs its activity and increases the size of the ubiquitin-D2 pool by 20- to 30-fold (97), suggesting that D2 ubiquitination is reversible, because not all Ub-D2 undergoes proteolysis. Enzymatic deubiquitination of ubiquitin-D2 occurs in vitro (164) and could explain recycling in vivo. D2 was recently identified as a substrate for the deubiquitinating enzymes VDU1 and VDU2 (165). Confocal studies indicate that both VDUs colocalize with D2, itself an integral endoplasmic reticulum membrane protein. The physical colocalization of VDU with D2 provides the opportunity for deubiquitination of D2.
VDU1-catalyzed D2 deubiquitination is an important part of the adaptive mechanism that regulates thyroid hormone action. In stimulated brown fat tissue, D2 increases intracellular T3 production, resulting in isolated tissue thyrotoxicosis (166,167,168). This is an important mechanism for cold acclimatization in rodents; mice with targeted inactivation of the D2 gene develop hypothermia and marked weight loss during cold exposure due to impaired thermogenesis in BAT (167,169). Increased VDU1-catalyzed deubiquitination of ubiquitin-D2, and therefore rescue of D2 from proteasomal degradation, is an integral part of this mechanism. In BAT, VDU1 mRNA levels are markedly upregulated by cold exposure or norepinephrine, which amplifies the transcriptional increase in D2 activity, and hence T3 production increases by approximately 2.5-fold. Although ubiquitination is known to play a physiologic role in several cellular processes (170,171,172,173,174), enzyme reactivation due to deubiquitination is unusual.
Related posts:
 Anatomy and Pathology of the Thyrotrophs
Anatomy and Pathology of the Thyrotrophs
 Iodine Deficiency and Endemic Cretinism
Iodine Deficiency and Endemic Cretinism
 Toxic Adenoma and Toxic Multinodular Goiter
Toxic Adenoma and Toxic Multinodular Goiter
 Thyroid Hormones in Thermogenesis, Intermediary Metabolism, and Obesity
Thyroid Hormones in Thermogenesis, Intermediary Metabolism, and Obesity
 Overview of the Clinical Manifestations of Hypothyroidism
Overview of the Clinical Manifestations of Hypothyroidism
 Hypothyroidism in Infants and Children: Neonatal Screening
Hypothyroidism in Infants and Children: Neonatal Screening
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


