Integrín αIIbβ3 and Platelet Aggregation
Ana Kasirer-Friede
Kamila Bledzka
Jun Qin
Edward F. Plow
Sanford J. Shattil
PERSPECTIVE
In 1977, Phillips and Agin1 provided important insight into the hemostatic function of platelets by demonstrating that two major glycoproteins, subsequently named IIb (GP IIb) andβ3 (GP IIIa), were missing from platelets of patients with Glanzmann thrombasthenia, a rare inherited bleeding disorder characterized by the absence of platelet aggregation. Earlier, Nurden and Caen2 had observed an abnormal gel pattern of membrane proteins of thrombasthenic platelets. Subsequently, it was found that αIIb and αIIb are subunits of a noncovalent complex, αIIbβ3.3,4 A second inherited disorder, afibrinogenemia, provided additional insight into the mechanisms of platelet aggregation. Platelets from patients with afibrinogenemia5 or washed normal platelets suspended in defibrinated plasma6 fail to aggregate; readdition of fibrinogen to the samples restored their aggregation response. In the late 1970s and 1980s, the connection between these observations was established when fibrinogen was found to associate with ADP-stimulated platelets,7 bind specifically to αIIbβ3,8,9,10 and mediate platelet aggregation.10,11 In the mid-1980s, another series of observations impacted on our understanding of the biology of αIIbβ3when it was discovered that αIIbβ3is a member of a widely distributed family of structurally, immunologically, and functionally related adhesion receptors,12,13,14 the integrins, so called because they integrated their extracellular ligands with the intracellular cytoskeleton.15 Thus, discoveries from studies of one integrin apply broadly to other family members.
These seminal observations have provided the underpinnings for investigations in many laboratories into the structure of αIIbβ3 and its function in platelet adhesion, aggregation, and hemostasis. Molecular details of how fibrinogen and other ligands interact with αIIbβ3 to effect platelet aggregation and adhesion have been defined. This work ultimately led to the development of parenteral αIIbβ3 antagonists for use in the prevention and treatment of arterial thrombosis in specific clinical settings.16 More recently, it has become apparent that integrins, including αIIbβ3, function not only as adhesion receptors but also as conduits for inside-out signals to regulate ligand binding to their extracellular domains and outside-in signals to regulate anchorage-dependent cellular responses, which in platelets include full aggregation and granule secretion as well as clot retraction. This chapter focuses on αIIbβ3 and its role in platelet aggregation and signaling.
EXPRESSION AND BIOSYNTHESIS OF αIIbβ3
αIIbβ3 synthesis is restricted primarily to megakaryocytes and consequently protein expression is limited to these cells and to platelets. However, αIIb expression has also been detected on early hematopoietic progenitors and on mast cells where in the latter it plays a role in adhesion and in the response of these cells to chronic inflammation.17,18 Restricted αIIbβ3expression is imposed by the αIIb-subunit because its promoter limits the expression to these hematopoietic cells.19,20 This is not the case for β3 since the integrin αvβ3 is expressed by many cell types, including platelets, endothelial cells, and smooth muscle cells. The genes for αIIb and β3 reside in close physical proximity on chromosome 17.21 αIIb and β3 must associate in a 1:1 noncovalent complex for efficient cell-surface expression,22 and naturally occurring mutations that alter the biosynthesis of one subunit affect surface expression of the other (e.g.,23,24,25). The association of nascent αIIb and β3 occurs in the endoplasmic reticulum.24,26 Although αIIb is synthesized as a single polypeptide chain, it is proteolytically cleaved into heavy and light chains by a furin-like enzyme in the Golgi complex. However, proteolytic cleavage is not required for αIIbβ3 expression on the cell surface nor is it required for αIIbβ3 function.27 αIIbβ3 is the most abundant cell-surface glycoprotein on platelets, present at approximately 80,000 copies per cell.28,29 In addition, an intracellular pool of αIIbβ3 is present in the membranes of α-granules and can become surface expressed upon granule secretion, increasing the number of functional αIIbβ3 receptors by approximately 30% to 50%.28,30,31 Rapid shuttling of αIIbβ3 between the cell surface and intracellular pools has been observed32 and may be involved in the import of fibrinogen into platelet α-granules.33
STRUCTURE OF αIIbβ3
αIIb and β3 consist of 1,008 and 762 amino acids, respectively34,35 The extracellular domains of both subunits are glycosylated.36 As assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis under nonreducing conditions, the apparent molecular weights of the αIIb- and β-subunits are approximately 130 kDa and 95 kDa, respectively22 Upon reduction, the αIIb-subunit separates into heavy and light chains with molecular weights of 115 and 25 kDa, respectively. The αIIb light chain contains a 20-amino acid cytoplasmic tail (CT), a transmembrane (TM) region, and an extracellular segment that is disulfide linked to the heavy chain. The β-subunit is a single polypeptide chain and contains 56 cysteine residues that form an extensive array of intrachain disulfide bonds.4 A schematic model depicting the domain organization of αIIbβ3 is shown in FIGURE 31.1. There are 16 domains in αIIbβ3:6 in αIIb and 10 in β3, including their TM and CT domains.
Structure of αIIb
The N-terminus of αIIb consists of seven approximately 60-amino acid repeats that fold to form a seven-bladed β–propeller. At the base of the β-propeller are four divalent cation-binding
motifs in which oxygenated residues within short hairpin loops coordinate divalent cations. The remainder of the αIIb extracellular region consists of a “thigh” and two “calf” domains. Between the thigh and the first calf domain is a “genu,” a flexible segment that allows the molecule to bend and compact. The αIIb TM domain is primarily helical.37,38 Two available nuclear magnetic resonance (NMR) structures of the TM domain differ in the vicinity of the inner-membrane leaflet37,38: one indicates that the helix is followed by an inverse turn, the other indicates a continuous helix. These differences will be important to resolve since several regulators of αIIbβ3 function are purported to bind to this region of αIIb39 Similarly, there are at least two different NMR structures for the αIIb CT In one structure, the membrane proximal region of the CT is composed of a short helix which terminates in a turn midway in the αIIb CT and an acidic C-terminus.40,41 A cation-binding site is present in this acidic C-terminal region, and occupancy of this site stabilizes the folding of the αIIb CT40,42 In a second structure, the region following the membrane proximal helix is intrinsically disordered, consistent with a lack of hydrophobic residues to facilitate folding.43
motifs in which oxygenated residues within short hairpin loops coordinate divalent cations. The remainder of the αIIb extracellular region consists of a “thigh” and two “calf” domains. Between the thigh and the first calf domain is a “genu,” a flexible segment that allows the molecule to bend and compact. The αIIb TM domain is primarily helical.37,38 Two available nuclear magnetic resonance (NMR) structures of the TM domain differ in the vicinity of the inner-membrane leaflet37,38: one indicates that the helix is followed by an inverse turn, the other indicates a continuous helix. These differences will be important to resolve since several regulators of αIIbβ3 function are purported to bind to this region of αIIb39 Similarly, there are at least two different NMR structures for the αIIb CT In one structure, the membrane proximal region of the CT is composed of a short helix which terminates in a turn midway in the αIIb CT and an acidic C-terminus.40,41 A cation-binding site is present in this acidic C-terminal region, and occupancy of this site stabilizes the folding of the αIIb CT40,42 In a second structure, the region following the membrane proximal helix is intrinsically disordered, consistent with a lack of hydrophobic residues to facilitate folding.43
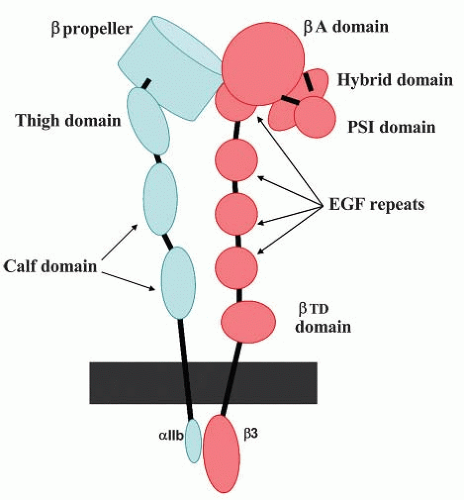 FIGURE 31.1 Schematic model showing the organization and structural domains of αIIbβ3. The α-subunit is in blue and the β-subunit is in red. PSI, ple×xin-semaphorin-integrin; EGF, epidermal growth factor, jβ-TD. |
Structure of β3
The N-terminus of β3 consists of two tandem nested domains. The first is the βA-domain whose fold is homologous to the I-domains present in the α-subunits of β2-containing integrins, as well as the α-subunits α1, α2, α10, α11, and αE (see FIGURE 31.1).44 The βA-domain is inserted in the “hybrid” domain whose fold is similar to I-set Ig domain. The hybrid domain is itself inserted in the PSI (plexin-semaphorin-mtegrin) domain that contains the N-terminus of β3. The C-terminus of the PSI domain is contiguous with a protease-resistant region composed of a series of disulfide repeats arranged into four EGF-like domains. The final structural motif in the β3 ectodomain is the βTD which extends to the outer leaflet of the plasma membrane.
The β3A-domain contains three divalent cation-binding sites: a metal ion-dependent adhesion site (MIDAS) motif that is involved in ligand binding, a site adjacent to MIDAS termed ADMIDAS that when occupied by Ca2+ inhibits ligand binding, and a ligand-induced metal-binding site, also known as the synergistic metal ion-binding site or SyMBS.45 The β3Adomain is folded into an α-β-α sandwich in which six central β-sheets are surrounded by seven α-helices. A loop on its upper surface termed the “specificity-determining loop” or SDL, in conjunction with a “cap” composed of four loops on the upper surface of αIIb β-propeller, forms a surface for ligand binding. The C-terminus of the PSI domain is contiguous with a protease-resistant region composed of a series of disulfide repeats arranged into four EGF-like domains. The final structural motif in the β3 ectodomain is the βTD which extends to the outer leaflet of the plasma membrane.
The β3 TM domain is a typical α-helix that extends into the CT. The helix is followed by a loop containing a kink,38,46,47 a turn, and then by another short helix.46 Noteworthy features of the β3 CT are two NXXY motifs, NPLY747 and NITY759. In general, NXXY motifs bind proteins containing phosphotyrosine binding (PTB) domains and this is also true of the two motifs in αIIbβ3.48,49 The interaction of these NXXY motifs with the PTB domains of binding partners has been implicated in regulating αIIbβ3 activation. The significance of the distal helix is demonstrated by the effects of the naturally occurring S752P mutation invariant Glanzmann thrombasthenia which disrupts the C-terminal helix, prevents binding of the kindlin-3 FERM domain to the β3 CT, and impairs αIIbβ3 activation.50,51
Interdomain and Intersubunit Interactions of αIIbβ3
Each of the major regions of αIIb and β3, that is, the ectodomains, the TM helices, and the CTs, interact with each other to establish an inactive heterodimeric structure. While there are no disulfide bonds between the integrin α– and β-subunits, intra- and interdomain disulfide bonds play a significant role in maintaining the overall conformation of the integrin.45,52,53
The binding sites for fibrinogen and other adhesive ligands are located within a globular head formed by the interaction of the β-propeller in αIIb-with the β3 A-domain.54 Chemical crosslinking,55,56 synthetic peptides,57,58,59 and site-directed antibodies60 have identified specific sequences as ligand-binding sites within αIIbβ3. Recent crystal structures of the αvβ53,61 and the αIIbβ3 extracellular domains complexed with peptides and small-molecule ligands54 have further refined the location of the binding sites. Thus, fibrinogen binds at the interface between the αIIb β-propeller and the β3A-domain with β3 contacts that include not only the MIDAS, but also a water-mediated interaction with the ADMIDAS, and with αIIb contacts in a groove between loops that connect the 2nd and 3rd and 3rd and 4th blades of the propeller. It is noteworthy that some of the drugs that inhibit ligand binding to αIIbβ3 do so through allosteric mechanisms, that is, they change the conformation of the receptor, rather than binding directly at the site that engages macromolecular ligands.54
Two smaller discontinuous interactions between α– and β-subunits in the stalk and between the head and the stalk further stabilize the heterodimeric structure of the inactive ectodomain.54,62 The α– and β-subunit TM helices also interact with each
other through multiple residues and cross at a 25- to 30-degree angle.37,38 The membrane-proximal portions of CTs of the two subunits also interact weakly through electrostatic and hydrophobic bonds.41,50 On the basis of mutagenesis experiments, it has been proposed that a salt bridge formed between αIIb residue R995 and β3 residue D723 is critical for maintaining αIIbβ3 in an inactive state,63 but a structure of sufficiently high resolution to directly visual the salt bridge is lacking. Nonetheless, based on mutational analyses,37,38 it is clear that changes in the interactions between the αIIb and β3 TM and CT domains regulate the ability of αIIbβ3 to bind extracellular matrix ligands.
other through multiple residues and cross at a 25- to 30-degree angle.37,38 The membrane-proximal portions of CTs of the two subunits also interact weakly through electrostatic and hydrophobic bonds.41,50 On the basis of mutagenesis experiments, it has been proposed that a salt bridge formed between αIIb residue R995 and β3 residue D723 is critical for maintaining αIIbβ3 in an inactive state,63 but a structure of sufficiently high resolution to directly visual the salt bridge is lacking. Nonetheless, based on mutational analyses,37,38 it is clear that changes in the interactions between the αIIb and β3 TM and CT domains regulate the ability of αIIbβ3 to bind extracellular matrix ligands.
PLATELET AGGREGATION IS MEDIATED αIIbβ3
The primary function of αIIbβ3 is to serve as a receptor for macromolecular ligands22,64 including fibrinogen, fibrin, von Willebrand factor (vWF), fibronectin, vitronectin, and thrombospondin (reviewed in65). All of these ligands are constituents of extracellular matrices where their interactions with αIIbβ3 can support platelet adhesion under a variety of conditions.66,67,68 Each ligand can also interact with at least one additional receptor on platelets. For example, fibrinogen can bind to αvβ369 and α5β1,70 vWF to GPIb/IX/V,71 fibronectin to α5β1,72 vitronectin to αvβ3,73 and thrombospondin to GPIV (CD36).74,75 Platelet aggregation requires engagement of αIIbβ3 by soluble fibrinogen8,10,11 and/or vWF.68 At high shear rates, such as those attained in stenosed arteries, vWF binding to αIIbβ3 is necessary for productive platelet aggregation.68,76,77,78
Ligand Recognition Speci.ficity of αIIbβ3
Fibrinogen binding to activated αIIbβ3 is divalent cation dependent, rapid, and initially reversible, with a dissociation constant (Kd) of approximately 300 nM.9,79,80,108 Stabilization then occurs, and the bound fibrinogen becomes essentially nondissociable.9,79,80,81,82,83 This transition stabilizes platelet aggregates and presumably depends upon the formation of additional contacts between fibrinogen and αIIbβ3.81,84 Stabilization maybe promoted by multiple factors, including ligand-induced structural changes in the receptor,82,85 receptor clustering, cytoskeletal reinforcements,86 fibrinogen self-association, association of fibrinogen with other adhesive proteins, such as thrombospondin,87,88 and receptor internalization.89 Although the dimeric structure of fibrinogen and the multimeric structure of vWF may permit these ligands to bridge between receptors on adjacent platelets and thereby mediate aggregation,90 such cross-bridging may not be the only mechanism that determines the size of platelet aggregates.91,92 Postreceptor occupancy events occur upon binding of fibrinogen to αIIbβ393 and are important for maximal aggregation. One direct manifestation of receptor occupancy is the induction of ligand-induced binding sites, which serve as epitopes for monoclonal antibodies that react with the occupied, but not with the resting or the activated but unoccupied conformers of αIIbβ3.94,95,96 Occupancy of αIIbβ3 also leads to a series of intracellular responses, referred to as outside-in signaling (see section on Regulation of Platelet Function by αIIbβ3.97,98,99
Two synthetic peptides of four to six amino acids define the recognition specificity of fibrinogen for αIIbβ3.100 The first of these peptides corresponds to the extreme C-terminus of the fibrinogen 7 chain.101 The C-terminal six amino acids of the γ chain are KQAGDV, and peptides corresponding to this sequence, or variants thereof, collectively referred to as the 7 chain peptides, interact with αIIbβ3.102 This sequence is present in fibrinogen but not in other αIIbβ3 ligands. Nevertheless, the /chain peptide does inhibit the binding of other adhesive proteins to αIIbβ3.91,103 A naturally occurring minor fibrinogen variant (the γ variant) that results from alternative splicing of the 7 chain mRNA alters the C-terminal sequence of the 7 chain and neither binds to αIIbβ3 nor supports platelet aggregation.104,105 Further, mutational studies conducted in vitro106 and in vivo107 confirm that the native/chain sequence is essential for the productive interaction of fibrinogen with αIIbβ3.
The second peptide, referred to as the RGD peptide, has Arg-Gly-Asp-X (where × includes most amino acids) as its minimal sequence. Two RGD sequences are present in the ha chain of human fibrinogen, and RGD-containing peptides inhibit fibrinogen binding to αIIbβ3 and platelet aggregation.108,109 Indeed, several of the αIIbβ3 therapeutic antagonists were designed from the RGD sequence.16,110 However, mutation of either RGD motif in the fibrinogen α chain does not alter fibrinogen recognition by the receptor.106 However, the RGD motifs that are present in several other αIIbβ3 ligands mediate their interaction with aαIIbβ3. The RGD motifs that are present in vWF111 and vitronectin112 are required to enable these proteins to interact with αIIbβ3. Fibronectin contains an RGD sequence as well as several alternative sequences that interact with αIIbβ3.113,114 RGD functions as a major recognition sequence for several other integrins, including αvβ3.115 Secondary roles for the RGD or other sequences in stabilizing αIIbβ3 interactions with its ligands have not been excluded. Roles for still other sequences, unrelated to the RGD and γchain peptides, in the αIIbβ3-dependent retraction of fibrin clots by platelets have also been demonstrated.116,117
In contrast to αIIbβ3, there are only a few hundred molecules of αIIbβ3 per platelet,118 and its physiologic significance is unclear. αIIbβ3 and αvβ3 share the same β-subunit, and their α-subunits are approximately 40% identical. Glanzmann thrombasthenia can arise from mutations in either the αIIb or β3 genes. Thus, β3 mutations produce αv*β3 as well as αIIbβ3 deficiency.23 However, phenotypic differences have not been reported between individuals lacking one or both β3 integrins. αIIbβ3 and αvβ3 bind many of the same ligands, including the two ligands that can mediate platelet aggregation, fibrinogen, and vWF,119 but ligands that bind to one receptor and not the other have also been identified; for example, osteopontin120 and adenovirus penton base121 bind to αvβ3 but not to αIIbβ3, whereas the snake venom barbourin binds to αIIbβ3 but not to αvβ3.122 This pattern is recapitulated at the level of the fine recognition specificities of the two integrins; RGD peptides bind with much higher affinity to αvβ3 than to αIIbβ3, and γ chain peptides bind with much higher affinity to αIIbβ3 than to αvβ3.123 Like αIIbβ3, αv*β3 can undergo activation,124,125,126,126 even in platelets.127 The breadth of functions ascribed to αvβ3 in cells other than platelets, including endothelial cells, has stimulated the development of specific αvβ3 antagonists, primarily for use as antiangiogenic drugs which would be useful in inhibiting tumor growth and metastasis,128 as well as for other diseases in which angiogenesis plays a prominent role.
αIIbβ3 Antagonists
Platelet adhesion is mediated by many receptors and platelet aggregation can be induced by many agonists, but aggregation itself is absolutely dependent upon αIIbβ3. This funneling of function onto αIIbβ3 provided a firm rationale for blockade
of its ligand-binding activity as an antithrombotic strategy and stimulated an intensive effort to develop specific and potent αIIbβ3 antagonists throughout the 1990s. In some respects, this effort was successful, but the hope that these drugs would provide both short-term and long-term protection against cardiovascular disease proved to be overly optimistic.129 Three αIIbβ3 antagonists, abciximab (ReoPro), eptifibatide (Integrilin), and tirofiban (Aggrastat), were approved by the US Food and Drug Administration as intravenously administered antithrombotics for specific indications. These drugs continue to be utilized to prevent acute coronary events in association with percutaneous coronary interventions (PCIs).130 Abciximab, the first αIIbβ3 antagonist approved for human use, is an Fab fragment of a humanized mouse monoclonal antibody to αIIbβ3.110,131,132 Abciximab also reacts with αvβ3 and αωβ2, and some of the positive effects of the drug may be attributable to this crossreactivity.133 Integrilin contains a K(Lys)GD within a structure rigidified within a disulfide loop.122 The RGD peptide was the starting point for tirofiban, but ultimately peptide bonds were designed out of its structure.134 Following on the early successes of these drugs in the setting of PCI, it was anticipated that they would also be efficacious in reducing acute coronary syndromes in general. However, clinical trials demonstrated only modest or no benefits, and ultimately they did not measure up well against other platelet antagonists, such as clopidogrel, in terms of cost and efficacy. Despite the anticipation that orally active αIIbβ3 antagonists would provide a long-term benefit to reduce the risk of thrombotic events,135 in clinical trials, these drugs were found at best to have little clinical benefit and, at worst, to be detrimental.136 As a result, the development of oral αIIbβ3 antagonists was abandoned. Thus, although the principle that αIIbβ3 blockade should prevent thrombus formation is mechanistically sound, the therapeutic window is narrow, meaning that underdosing would be ineffective and overdosing could cause bleeding. At this juncture, parenteral αIIbβ3 antagonists are only used for limited indications.129 The development of inhibitors that directly target αIIbβ3 continues to be explored. Antagonists that block ligand binding without engaging the β3 MIDAS metal ion or disrupt interactions of the β3 CT with its activating binding partners may be the targets of future antithrombotic drugs.98,137
of its ligand-binding activity as an antithrombotic strategy and stimulated an intensive effort to develop specific and potent αIIbβ3 antagonists throughout the 1990s. In some respects, this effort was successful, but the hope that these drugs would provide both short-term and long-term protection against cardiovascular disease proved to be overly optimistic.129 Three αIIbβ3 antagonists, abciximab (ReoPro), eptifibatide (Integrilin), and tirofiban (Aggrastat), were approved by the US Food and Drug Administration as intravenously administered antithrombotics for specific indications. These drugs continue to be utilized to prevent acute coronary events in association with percutaneous coronary interventions (PCIs).130 Abciximab, the first αIIbβ3 antagonist approved for human use, is an Fab fragment of a humanized mouse monoclonal antibody to αIIbβ3.110,131,132 Abciximab also reacts with αvβ3 and αωβ2, and some of the positive effects of the drug may be attributable to this crossreactivity.133 Integrilin contains a K(Lys)GD within a structure rigidified within a disulfide loop.122 The RGD peptide was the starting point for tirofiban, but ultimately peptide bonds were designed out of its structure.134 Following on the early successes of these drugs in the setting of PCI, it was anticipated that they would also be efficacious in reducing acute coronary syndromes in general. However, clinical trials demonstrated only modest or no benefits, and ultimately they did not measure up well against other platelet antagonists, such as clopidogrel, in terms of cost and efficacy. Despite the anticipation that orally active αIIbβ3 antagonists would provide a long-term benefit to reduce the risk of thrombotic events,135 in clinical trials, these drugs were found at best to have little clinical benefit and, at worst, to be detrimental.136 As a result, the development of oral αIIbβ3 antagonists was abandoned. Thus, although the principle that αIIbβ3 blockade should prevent thrombus formation is mechanistically sound, the therapeutic window is narrow, meaning that underdosing would be ineffective and overdosing could cause bleeding. At this juncture, parenteral αIIbβ3 antagonists are only used for limited indications.129 The development of inhibitors that directly target αIIbβ3 continues to be explored. Antagonists that block ligand binding without engaging the β3 MIDAS metal ion or disrupt interactions of the β3 CT with its activating binding partners may be the targets of future antithrombotic drugs.98,137
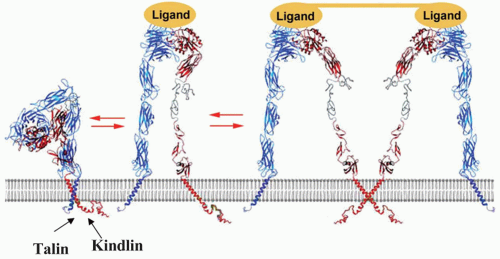 FIGURE 31.2 Model of a pathway for activation of αIIbβ3. Talin and kindlin binding to the CT of the β3-subunit can lead to unclasping of integrin subunit tails and a membrane-associated structural change of the cytoplasmic face. This change may entail a transition from a bent conformation in its resting state to an extended conformation in which it becomes competent to bind soluble ligands.The α-subunit is in blue and the β-subunit is in red. (Reproduced from Qin J, Vinogradova O, Plow EF. Integrin bidirectional signaling: a molecular view. PLoS Biol 2004;2:e169, with permission.) |
REGULATION OF αIIbβ3 ACTIVATION (INSIDE-OUT SIGNALING)
Key to the ability of αIIbβ3 to bind fibrinogen and vWF and support platelet aggregation is its ability to undergo a transformation from a resting low-affinity state to an activated high-affinity state as depicted in FIGURE 31.2. The overall shape of αIIbβ3 depicted in this figure is based on cryogenic electron micrographic images of the isolated receptor,138 crystal structures of the homologous αvβ3 extracellular domain (Protein Data Bank: 1JV2,1M1X)53,61 and the αIIbβ3 extracellular domain (Protein Data Bank 3FCS),45 as well as NMR analyses of the TM and CT (Protein Data Bank 1M80)41,46 (Protein Data Bank 2K9J).37,38,139 In this model, the globular head of resting, unactivated αIIbβ3 rests on two stalks. By bending at the genu of the αIIb and β3 ectodomains, the head approaches the cell membrane140,141 (see FIGURE 31.2). When αIIbβ3 is activated, its height increases as its legs separate and straighten.142 In association with leg separation, one of the helices in the β3A-domain and the hybrid domain swings away to render the ligand-binding site formed between the β-propeller in the αIIb-subunit and the β3A- domain accessible to macromolecular ligands.142,143,144 While these
changes reflect the behavior of the ectodomains of the majority of αIIbβ3 molecules on the platelet surface during activation, it appears that some αIIbβ3 molecules are capable of binding ligands while still in a bent or partially unfolded conformation,145 implying that extension of the ectodomain may also occur as a postactivation event.146 The inside-out signals that induce these activation events may also promote clustering of αIIbβ3 heterodimers into heterooligomers147; clustering would be expected to increase αIIbβ3 avidity or valency148 However, conformational change and affinity modulation appear to be the dominant mechanism by which the ligand-binding activity of αIIbβ3 (and αvβ3) is regulated.125,149
changes reflect the behavior of the ectodomains of the majority of αIIbβ3 molecules on the platelet surface during activation, it appears that some αIIbβ3 molecules are capable of binding ligands while still in a bent or partially unfolded conformation,145 implying that extension of the ectodomain may also occur as a postactivation event.146 The inside-out signals that induce these activation events may also promote clustering of αIIbβ3 heterodimers into heterooligomers147; clustering would be expected to increase αIIbβ3 avidity or valency148 However, conformational change and affinity modulation appear to be the dominant mechanism by which the ligand-binding activity of αIIbβ3 (and αvβ3) is regulated.125,149
Substantial progress has been made in understanding how integrin activation and the consequent movements of the ectodomain are triggered. In platelets, agonist signaling (inside-out signaling) eventuates in protein binding to and alterations in the interactions of the resting αIIbβ3 CT domain complex. A PTB domain located in the 270 kDa cytoskeletal protein talin binds to the β3 CT domain and disrupts the hydrophobic and electrostatic bonds that maintain it in complex with the αIIb CT domain. In turn, dissociation of this CT clasp affects insertion of the β3 CT domain into the platelet plasma membrane and disrupts the association between the αIIb and β3 TM helices. Disengagement of the TM helices then provides the force needed to trigger the activating events in the αIIbβ3 ectodomain. 37,38,41,139,140,150-152 0nce the CT and TM domains have dissociated, the propensity of the subunits to homooligomerize may also increase to influence the activation process.147 With the ready availability of αIIbβ3 ligands in plasma, tight control of αIIbβ3 activation is essential. This regulation is achieved through the binding of cytosolic proteins to the α– and β-subunit CT domains. For integrin β CT, approximately 40 binding partners have been identified.153 Among these, talin, filamin, kindlin, and integrin-linked kinase (ILK) are known to influence αIIbβ3 activation. Exactly how each does so remains unclear in many cases. Some of the proteins appear to induce or enhance integrin activation while others act as negative regulators.37,154,155,156
Proximal Regulation of αIIbβ3 Activation
A schematic identifying some of the intermediates in αIIbβ3 activation is shown in FIGURE 31.3. Talin binds directly to the CT of αIIbβ3 and is essential for integrin activation.157 Talin is composed of an approximately 50 kDa head domain (talin-H) and a approximately 220 kDa rod domain (talin-R). The talin-H resembles a FERM (4.1 ezrin radixin moesin) domain and contains F1-, F2-, and F3-PTB subdomains.158 In the resting platelet, talin exists in an autoinhibited conformation in which the talin-H interacts with talin-R.159,160 The β3 CT-binding site in talin-H can be exposed by calpain cleavage,161 RIAM (Rap 1 interacting adaptor molecule) association,162 or phosphoinositide binding.163 Once unfolded, talin can interact with the αIIbβ3 CT.155,164 The talin-H binds to two sites in the β3 CT: the membrane-proximal NPLY747 motif with high affinity165 and a membrane proximal region near the plasma membrane41,166 with lower affinity. It is the second interaction that disrupts the clasp between the β3 CT and the αIIb CT, triggering the activation process. Talin-R also contains an integrinbinding site.167
Although it is clear that talin is necessary, and in model cell systems sufficient, to induce at least partial integrin activation,155,168,169 in mice170,171 and humans,172,173 at least one additional molecule is essential for robust integrin activation. In platelets and other hematopoietic cells, this molecule is kindlin-3, and in other cells, the other two kindlin family members, kindlin-1 and kindlin-2, cooperate with talin to fulfill this coactivator role.174,175,176,177,178 The coactivator role was originally demonstrated for kindlin-2, where the combination of kindlin-2 with talin-H-induced robust activation of integrins compared to talin-H alone.168,179 The three mammalian kindlins have molecular weights of approximately 75 kDa. Like talin-H, kindlins also have a FERM domain. In kindlins, the F2 subdomain is interrupted by a pleckstrin homology (PH) domain. Four kindlin-binding partners have been identified: ILK,179,180,181
migfilin,181,182 and β-integrin and/33-integrin CT.179,183,184 Like talin, kindlins bind to β CT through their F3-PTB, but unlike talin, they bind to the membrane distal NXXY759 motif (NITY in β3).168,185 Phosphorylation of the Y in this sequence dissociates kindlin from the β3 CT.186 Kindlin-3 deficiency in humans gives rise to the leukocyte adhesion deficiency-III syndrome, LAD-III, also referred to as integrin activation deficiency disease173 that is characterized by episodic bleeding of variable severity, increased susceptibility to infections, and sometimes osteopetrosis172,173 and results from an inability to activate both β2– and β3-containing integrins. ILK, a 59 kDa cytoplasmic protein, can interact with the cytoplasmic domain of βv β2, and β3 integrin subunits.187 Deficiency in mice or knockdown of ILK leads to defective platelet function,188,189 although not as severe as that associated with kindlin-3 deficiency.
migfilin,181,182 and β-integrin and/33-integrin CT.179,183,184 Like talin, kindlins bind to β CT through their F3-PTB, but unlike talin, they bind to the membrane distal NXXY759 motif (NITY in β3).168,185 Phosphorylation of the Y in this sequence dissociates kindlin from the β3 CT.186 Kindlin-3 deficiency in humans gives rise to the leukocyte adhesion deficiency-III syndrome, LAD-III, also referred to as integrin activation deficiency disease173 that is characterized by episodic bleeding of variable severity, increased susceptibility to infections, and sometimes osteopetrosis172,173 and results from an inability to activate both β2– and β3-containing integrins. ILK, a 59 kDa cytoplasmic protein, can interact with the cytoplasmic domain of βv β2, and β3 integrin subunits.187 Deficiency in mice or knockdown of ILK leads to defective platelet function,188,189 although not as severe as that associated with kindlin-3 deficiency.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Molecular Basis for Platelet Secretion
Molecular Basis for Platelet Secretion
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


