Insulin Resistance and Its Role in the Pathogenesis of Type 2 Diabetes
Meredith Hawkins
Luciano Rossetti
The term insulin resistance generally refers to resistance to the metabolic effects of insulin, including the suppressive effects of insulin on endogenous glucose production, the stimulatory effects of insulin on peripheral (predominantly skeletal muscle) glucose uptake and glycogen synthesis, and the inhibitory effects of insulin on adipose tissue lipolysis.
Following its discovery by Banting and Best in 1921, insulin rapidly entered the clinical arena and dramatically extended the life spans of individuals with type 1 diabetes mellitus. Chronic treatment with exogenous insulin permitted the recognition of individuals with very high insulin requirements, indicating resistance to the hormone (1). Many such individuals had secondary causes for insulin resistance, including high titers of antibodies to insulin after chronic treatment with insulin from animal sources (2), as well as rare genetic defects in insulin signaling (3). However, the thriving of patients with type 1 diabetes into their later years and the treatment of individuals with type 2 diabetes with insulin revealed the phenomenon of insulin resistance secondary to obesity and aging, which was widely recognized by the 1970s (3). Many of the considerable advances of the past three decades in understanding the pathogenesis of insulin resistance will be reviewed below.
CLINICAL FEATURES AND SIGNIFICANCEOF INSULIN RESISTANCE
The clinical importance of insulin resistance cannot be overstated, given the mortality and morbidity associated with the many disorders that are likely a consequence of this condition. Indeed, there is considerable epidemiologic evidence linking insulin resistance with glucose intolerance and type 2 diabetes, hypertension, dyslipidemia, atherosclerosis, and many cancers (4,5,6,7,8). It is generally accepted that insulin resistance plays a major role in the development of type 2 diabetes (9). Because currently an estimated 170 million people worldwide have type 2 diabetes (10), this is clearly an important association. Indeed, prospective studies have revealed that insulin resistance predates the onset of type 2 diabetes by 10 to 20 years and is the best clinical predictor of subsequent development of type 2 diabetes (11,12). Furthermore, insulin resistance is a consistent finding in patients with type 2 diabetes (3,13,14,15). However, the development of frank diabetes mellitus appears to require an additional defect in insulin secretion (11). In the absence of a defect in β-cell function, individuals can compensate indefinitely for insulin resistance with appropriate hyperinsulinemia (11). Hence, many individuals with marked insulin resistance
may never progress to type 2 diabetes. However, the risk of atherosclerosis is apparently comparable in nondiabetic, insulin-resistant individuals and in those with type 2 diabetes (16).
may never progress to type 2 diabetes. However, the risk of atherosclerosis is apparently comparable in nondiabetic, insulin-resistant individuals and in those with type 2 diabetes (16).
Metabolic Syndrome
Reaven formally recognized the clinically significant association between insulin resistance and cardiovascular risk factors in his description of the syndrome of insulin resistance in 1988 (17). The most recent definition of the metabolic syndrome was established as a consensus by the National Cholesterol Education Program (NCEP) Adult Treatment Panel III (ATPIII) in 2001 (18). Based on the panel’s guidelines, diagnosis of the metabolic syndrome requires the presence of at least three of the following five criteria: elevated fasting plasma glucose levels (>110 mg/dL), visceral obesity (waist circumference >35 inches in women and 40 inches in men), hypertension (>130/85 mm Hg), hypertriglyceridemia (>150 mg/dL), and low high-density lipoprotein (HDL) cholesterol (<40 mg/dL in men and <50 mg/dL in women). By this definition, the estimated prevalence of the metabolic syndrome in the United States is currently greater than 20% among all adults older than 20 years of age, and greater than 40% among the population older than 50 (19).
Other recognized components of the syndrome include systemic inflammation, a prothrombotic state, and increased oxidant stress (20). The increased circulating levels of tumornecrosis factor-α (TNF-α), interleukin-6 (IL-6), and other proinflammatory cytokines may contribute to some of the metabolic features of the metabolic syndrome, whereas the increased levels of plasminogen activator inhibitor-1 (PAI-1) appear to heighten the risk of atherothrombosis (21). Indeed, an increased risk of atherosclerotic disease mortality and morbidity is conferred by the metabolic syndrome. Other associated clinical disorders include polycystic ovarian syndrome, nonalcoholic fatty liver disease, and sleep apnea (14).
Contributions of Tissue-Specific Insulin Resistance
Defects in the ability of insulin to stimulate skeletal muscle glucose uptake and to suppress hepatic glucose production and adipose tissue lipolysis all tend to coexist in insulin-resistant individuals with established type 2 diabetes (22). It has been speculated that the onset of insulin resistance in some tissues may precede the onset in others. Although it has been proposed that insulin resistance develops later in the liver than in skeletal muscle, hepatic glucose production is inappropriately unsuppressed in the presence of hyperinsulinemia during the development of insulin resistance (23). Additionally, marked hepatic insulin resistance precedes the onset of peripheral insulin resistance secondary to moderate fat feeding (24). Various experimental models have been developed to define organ-specific contributions of insulin resistance to hyperglycemia and other metabolic abnormalities.
SKELETAL MUSCLE
A universally recognized feature of type 2 diabetes is skeletal muscle insulin resistance, secondary to either genetic or metabolic factors (25). Insulin clamp studies have permitted the measurement of peripheral and hepatic glucose fluxes in the presence of fixed glucose and insulin levels by means of continuous intravenous infusions and frequent measurement of plasma glucose levels (26,27). These sophisticated physiologic measures have revealed that skeletal muscle is quantitatively the most important tissue involved in systemic glucose homeostasis, because it accounts for ∼80% of glucose disposal following glucose infusion or ingestion (11). Thus, decreased ability of insulin to stimulate glucose disposal by this tissue is of considerable importance to whole-body glucose homeostasis.
Indeed, impaired insulin-dependent glucose uptake and phosphorylation have been documented at early stages in the development of type 2 diabetes (28). Several transgenic mouse models have examined the impact of isolated defects in skeletal muscle insulin action on whole-body glucose intolerance. Conditional inactivation of the insulin receptor in skeletal muscle by Cre-mediated recombination gave rise to a model of the metabolic syndrome, with increased fat stores and hypertriglyceridemia (29). Despite the changes in fat metabolism, these muscle-specific insulin receptor knockout (MIRKO) mice did not develop hyperinsulinemia and diabetes, suggesting that there was adequate shunting of glucose utilization from muscle to adipose tissue to prevent excursions in blood glucose (30). Another model of severe skeletal muscle insulin resistance was generated by crossing mice heterozygous for a systemic insulin receptor knockout with mice bearing a dominant-negative insulin receptor transgene in muscle. Despite a greater than 90% decrease in insulin receptor kinase activity and reduced insulin-stimulated glucose uptake, these mice still did not develop diabetes (31).
The findings stand in sharp contrast to studies showing that ablation of the insulin-dependent glucose transporter GLUT4 in skeletal muscle can cause diabetes (32). There are several explanations for this apparent contradiction. In mice lacking the insulin receptor in muscle, both the contraction-activated pathway (33,34) and the insulin-like growth factor I (IGF-I) signaling pathway (35) can compensate for the ablation of insulin signaling. The former, which activates adenosine monophosphate (AMP)-activated protein kinase and thereby stimulates translocation of glucose transporters (36), remains intact in MIRKO mice. The importance of the IGF-I signaling system in muscle metabolism is highlighted by a mouse model of combined ablation of insulin and IGF-I receptor function in skeletal muscle (37). Unlike mice with isolated ablation of the insulin receptor in skeletal muscle, these mice develop frank diabetes and other metabolic features characteristic of insulin resistance. Together, these models confirm the central role of skeletal muscle as a site of insulin action, yet indicate that there are alternate pathways leading to glucose uptake and GLUT4 translocation in skeletal muscle that can compensate in mice lacking the insulin receptor.
Insulin-stimulated 3- O -methylglucose transport into isolated skeletal muscle from patients with type 2 diabetes is substantially lower than in normal controls, demonstrating that decreased skeletal muscle uptake contributes to impaired peripheral glucose uptake (36,38). A uniform finding in both obesity and type 2 diabetes is decreased insulin receptor substrate-1 (IRS-1)-associated tyrosine phosphorylation and 1-phosphatidylinositol 3-kinase (PI 3-kinase) activity in skeletal muscle (39,40,41). Of note, insulin action on glucose transport is normalized in isolated muscle strips from patients with type 2 diabetes after a 2-hour in vitro incubation in the presence of 5 mM glucose (42). Similarly, a more recent report provided evidence that proximal insulin signaling parameters elicit a normal response in cultured myotubes prepared from muscle biopsies from insulin-resistant nondiabetic subjects (43). These findings support the hypothesis that insulin resistance of skeletal muscle in individuals with type 2 diabetes is at least in part secondary to an altered metabolic milieu, as will be explored further under “Environmental” Factors Contributing to Insulin Resistance.
In addition to this downregulation of proximal insulin signaling, several negative regulators of insulin signaling are upregulated in insulin resistance. Plasma-cell differentiation factor-1 (PC-1) is a membrane glycoprotein with ectonucleotide
pyrophosphatase activity that seems to act as an intrinsic inhibitor of insulin receptor tyrosine kinase activity (44,45). In healthy subjects with no clinically significant defects in glucose metabolism, PC-1 expression in muscle is negatively correlated with insulin sensitivity and in vitro stimulation of muscle insulin receptor tyrosine kinase activity (46,47). It has been suggested that increased expression of PC-1 in skeletal muscle of obese subjects is more strongly associated with downregulation of insulin receptor tyrosine phosphorylation than with decreased insulin receptor expression, but this requires further investigation (48).
pyrophosphatase activity that seems to act as an intrinsic inhibitor of insulin receptor tyrosine kinase activity (44,45). In healthy subjects with no clinically significant defects in glucose metabolism, PC-1 expression in muscle is negatively correlated with insulin sensitivity and in vitro stimulation of muscle insulin receptor tyrosine kinase activity (46,47). It has been suggested that increased expression of PC-1 in skeletal muscle of obese subjects is more strongly associated with downregulation of insulin receptor tyrosine phosphorylation than with decreased insulin receptor expression, but this requires further investigation (48).
Phosphotyrosine phosphatases (PTPases) are enzymes that dephosphorylate the insulin receptor and its substrates, thereby turning off the insulin signal. Total membrane-bound tyrosine phosphatase activity is increased in skeletal muscle of patients with type 2 diabetes (49), particularly protein-tyrosine phosphatase-1B (PTP-1B) (50), which negatively regulates phosphorylation of the insulin receptor and IRS-1 (51). Mice with ablation of the PTP1B gene demonstrate increased insulin sensitivity and enhanced insulin-stimulated phosphorylation of the insulin receptor in both skeletal muscle and liver (52). These mice are also resistant to weight gain on a high-fat diet, demonstrating the potential importance of this enzyme in mediating the central effects of insulin on appetite, discussed under Brain. Selective reduction of PTP1B protein and messenger RNA (mRNA) in liver and fat normalized plasma glucose levels, reduced hyperinsulinemia, and reduced expression of gluconeogenic enzymes in genetically obese and diabetic mice (53). These promising in vivo rodent data have prompted a highly motivated search for pharmacologic inhibitors of this enzyme (54).
ADIPOSE TISSUE
Insulin resistance in adipose tissue is characterized by decreased suppression of adipose tissue lipolysis by insulin, resulting in elevated circulating levels of free fatty acids (FFAs). Indeed, the suppressive effect of insulin on FFA levels is impaired in obese insulin-resistant individuals (55,56) and in type 2 diabetes (57). The presence of the same defect in glucose-tolerant first-degree relatives of patients with type 2 diabetes (58,59) suggests that abnormal insulin-mediated suppression of plasma FFA is an early defect in those with a genetic predisposition to insulin resistance. Although it has been suggested that increased FFA levels in obese individuals are due primarily to expansion of body-fat depots (60,61), the suppressive effect of insulin on FFA levels is also reduced in nonobese insulin-resistant individuals (62).
Transgenic models of adipocyte-specific disruption of insulin signaling have been generated to address the impact of decreased adipocyte insulin action on systemic fat and glucose metabolism. Conditional ablation of the insulin receptor in white and brown adipocytes with an adipose-specific fatty acid binding protein aP2 promoter (63) caused a marked decrease in gonadal fat mass and whole-body triglyceride content. Moreover, these fat-specific insulin receptor knockout (FIRKO) mice are resistant to weight gain with aging or hypothalamic insult and are protected against hyperphagia-induced glucose intolerance. Since the aP2 promoter would also target macrophages, these mice may also have been spared the adverse metabolic effects of systemic inflammation, discussed below. Thus, although insulin signaling is required for triglyceride storage in adipocytes, it may not be essential for normal glucose metabolism.
By contrast, knockout of the insulin-dependent glucose transporter GLUT4 in fat induced hepatic and skeletal muscle insulin resistance and glucose intolerance (64). However, fasting serum FFA levels were suppressed appropriately in response to insulin in these knockout mice, suggesting that the insulin resistance was relatively selective for glucose uptake. Furthermore, these mice displayed normal muscle triglyceride content and normal levels of leptin and TNF-α. Thus, the insulin resistance observed in these mice may be secondary to chronic hyperinsulinemia or to altered secretion of adipocyte-derived molecules that mediate insulin action in other tissues (see below).
Ob/ob mice that lack the adipose tissue fatty acid binding protein aP2 have reduced adipose tissue lipolysis and increased adipose tissue mass, together with a paradoxical reduction in plasma lipids and an improvement in insulin sensitivity and insulin secretion (65). This suggests that enlargement of adipose tissue mass may have protective effects against insulin resistance. This concept of adipose tissue providing a “sink” to protect other tissues from the toxic effects of excessive fatty acids is supported by the observation that overexpression of GLUT4 in adipose tissue of transgenic mice causes both an increase in adipose tissue mass and an improvement in whole-body insulin sensitivity (66,67). Further evidence for beneficial effects of adipose tissue will be provided in the subsequent discussion of lipodystrophy and fatless mice and the reversal of insulin resistance with adipose tissue transplantation in those transgenic models.
LIVER
There has been considerable controversy about whether hepatic glucose production is directly regulated by insulin or is mediated via extrahepatic metabolic effects. In particular, it has been proposed that insulin suppresses glucose production by reducing flux of amino acids and FFA from muscle and adipose tissue to the liver, thereby also reducing gluconeogenesis (68,69,70,71,72). Normal inhibitory action of insulin on hepatic glucose production appears to require normal insulin responsiveness at the level of adipose tissue. Indeed, the ability of peripheral insulin to regulate glucose production appears to be at least in part due to its ability to suppress circulating FFA levels (73,74).
Evidence for a direct effect of insulin on hepatic glucose production is provided by a mouse model in which the insulin receptor is ablated in muscle and adipose tissue, with normal insulin signaling in the liver (75). These mice maintain normal hepatic insulin sensitivity and do not progress to diabetes despite impaired glucose tolerance, suggesting that hepatic insulin resistance is required for the onset of overt diabetes. This model may also suggest that insulin resistance in the liver is an intrinsic abnormality of insulin signaling in the hepatocyte. Indeed, mice with conditional, liver-specific knockout of the insulin receptor (LIRKO) exhibit marked insulin resistance, glucose intolerance, and an inability of insulin to suppress hepatic glucose production and regulate hepatic gene expression (76,77). Furthermore, the liver might actually affect peripheral insulin action. Some evidence for an association between hepatic fat accumulation and peripheral insulin resistance is presented below in the discussion of the metabolic effects of intrahepatocellular triglyceride.
BRAIN
Hypothalamic resistance to the central appetite-suppressing and metabolic effects of insulin may play seminal roles in the development of insulin resistance. The insulin receptor is widely expressed in several brain areas (78) and has been implicated in the regulation of satiety (79). Although glucose disposal in the majority of neurons occurs in an insulin-independent manner, neurons in the hypothalamus and other discrete brain areas express the insulin-responsive glucose transporter GLUT4 (80). The potential role of brain insulin signaling has been studied by
generating a neuron-specific insulin receptor knockout (NIRKO), resulting in increased food intake and moderate diet-dependent obesity (81).
generating a neuron-specific insulin receptor knockout (NIRKO), resulting in increased food intake and moderate diet-dependent obesity (81).
The metabolic effects of insulin action in the brain have recently been examined using intracerebroventricular injections of antisense oligonucleotides and blocking antibodies to the insulin receptor (82). These manipulations caused a targeted impairment in hypothalamic insulin receptor function in rats, with a rapid onset of hyperphagia and a significant increase in fat mass after only 7 days, similar to the NIRKO mouse. Of particular note, this central inhibition of insulin action also dramatically reduced the ability of exogenously infused insulin to blunt hepatic glucose output, demonstrating an important role for the hypothalamic insulin receptor in the regulation of hepatic glucose metabolism (83).
An Integrated Model of Insulin Resistance
As shown in Figure. 24.1, it is likely that several factors contribute to impaired insulin action in most individuals with insulin resistance. The following sections will discuss the interrelationship between genetic inheritance, obesity, and environmental factors in the pathogenesis of insulin resistance.
 Figure 24.1. Pathogenesis of insulin resistance: contributions of genes, obesity, and environment. “Environment” refers to a variety of factors, including hormones, increased nutrient availability, and age. |
GENETIC CAUSES OF INSULIN RESISTANCE
Defects in both insulin action and insulin secretion are present in type 2 diabetes, and both are believed to be genetically predetermined (84). A strong genetic basis for insulin resistance is suggested by the high prevalence in certain populations, particularly the Nauru Islanders of the Pacific (85), the Pima Indians in Arizona (86), and the urban Wanigela people in Papua New Guinea (87). Although the current epidemic levels of insulin resistance and type 2 diabetes in these populations have followed the introduction of a “Westernized” lifestyle characterized by high caloric intake and physical inactivity, their prevalence is much higher in these populations than in populations of other ethnic groups with the same lifestyle. Furthermore, there is a nearly 100% concordance in diagnosis of type 2 diabetes between monozygotic twins but only a 20% concordance between dizygotic twins (88). To date, documented monogenic inheritance of insulin resistance has been limited to a few families with extreme forms of the disorder, as described in the next section.
Extreme Insulin Resistance
To date, several rare mutations in genes associated with insulin action have been linked to extreme insulin resistance (Fig. 24.2). Although these are all rare conditions, their study has been helpful in elucidating issues pertinent to more general pathophysiology of insulin resistance. All of the following genetic conditions are associated with typical clinical manifestations: hyperinsulinemia, dyslipidemia, hypertension, and impaired glucose tolerance or insulin-resistant diabetes (89). Acanthosis nigricans is a characteristic skin lesion in insulin resistance, consisting of velvety and papillomatous pigmented hyperkeratosis of the flexures and neck (90). In women, extreme insulin resistance is also associated with hyperandrogenism, hirsutism, menstrual abnormalities, and polycystic ovarian disease.
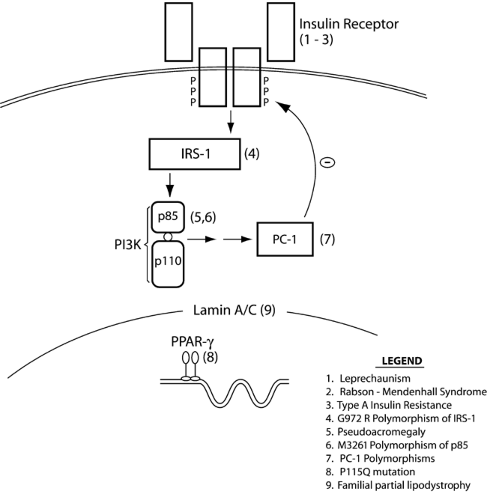 Figure 24.2. Genetic causes of insulin resistance identified in humans. This schematic representation depicts the cellular localization of proteins in which mutations or polymorphisms have been identified in association with extreme insulin resistance. In the case of the insulin receptor, multiple mutations have been identified that contribute to the syndromes indicated. Numerically, the mutations/polymorphisms/syndromes are as follows: (1) leprechaunism; (2) Rabson-Mendenhall syndrome; (3) type A insulin resistance; (4) G972R polymorphism of insulin receptor substrate-1 (IRS-1); (5) pseudoacromegaly; (6) M3261 polymorphism of p85; (7) Plasma-cell differentiation factor-1 (PC-1) polymorphisms; (8) P115Q mutation of peroxisome proliferator-activated receptor γ (PPARγ); (9) familial partial lipodystrophy (lamin A/C mutations). PI3K, 1-phosphatidyl-inositol 3-kinase. |
INSULIN RECEPTOR MUTATIONS
Severe insulin resistance has been reported in conjunction with more than 100 naturally occurring mutations in the insulin receptor gene (91,92), as well as in the complete absence of insulin receptors (93). The varying severity and diversity of their phenotypes have resulted in the description of several clinical syndromes of severe insulin resistance (94).
Leprechaunism is the most extreme form of these syndromes, characterized by intrauterine growth retardation and characteristic dysmorphic features including prominent eyes, thick lips, upturned nostrils, low-set posteriorly rotated ears, and thick skin with lack of subcutaneous fat (95). Life expectancy is extremely short, with death usually before 1 year of age. Milder syndromes of insulin resistance have been reported in patients whose insulin receptor mutations do not lead to a complete loss of insulin receptor function (96). Children with the somewhat milder Rabson-Mendenhall syndrome have life expectancies of up to 15 years and display different dysmorphic features, with premature or dysplastic teeth and gingival hyperplasia, as well as growth retardation. Despite postprandial hyperglycemia, affected children with either syndrome demonstrate fasting hypoglycemia due to inappropriately elevated fasting insulin levels. Type A insulin resistance is the mildest syndrome of insulin receptor gene mutations and is characterized by a classic triad of insulin resistance, acanthosis nigricans, and hyperandrogenism (in females) (97). Most patients with type A insulin resistance are heterozygous for a single mutant allele, most frequently a mutation in the tyrosine kinase domain of the receptor (98), and most such patients do not develop diabetes. However, patients with two mutant alleles tend to develop overt diabetes in childhood or adolescence (99).
The pronounced mitogenic features and fasting hypoglycemia in individuals with the above syndromes, despite the marked resistance to the metabolic effects of insulin, suggest that the high circulating insulin levels may be mediating these mitogenic effects through the homologous IGF-I receptor (100). Indeed, this receptor homology has been exploited therapeutically in the treatment of diabetes in these patients. Daily injections of IGF-I were effective in lowering both fasting and postprandial plasma glucose concentrations for up to 16 months in 11 patients with extreme insulin resistance (101). Even more dramatically, a recent report demonstrated that treatment of a patient with leprechaunism from infancy until 7 years of age with recombinant human IGF-I maintained her growth rate and hemoglobin A1C level nearly within the normal range (102).
INSULIN-MEDIATED PSEUDOACROMEGALY
A syndrome of severe insulin resistance has been described in which patients develop pathologic tissue growth similar in appearance to that of acromegaly but without elevated levels of growth hormone or IGF-I (103). Although neither insulin nor IGF-I can activate PI 3-kinase or stimulate glucose uptake in cultured skin fibroblasts from these individuals, mitogen-activated protein kinase (MAPK) phosphorylation and downstream mitogenic signaling by both hormones are intact. No defects have
been documented in the structure, expression, or activation of the insulin receptor, IRS-1, or p85α, suggesting a selective defect of postreceptor insulin signaling to metabolic pathways. The acromegaloid tissue growth is likely due to activation of the intact mitogenic signaling pathways by the severe hyperinsulinemia (104).
been documented in the structure, expression, or activation of the insulin receptor, IRS-1, or p85α, suggesting a selective defect of postreceptor insulin signaling to metabolic pathways. The acromegaloid tissue growth is likely due to activation of the intact mitogenic signaling pathways by the severe hyperinsulinemia (104).
MUTATIONS IN THE PEROXISOME PROLIFERATOR-ACTIVATED RECEPTOR γ
The nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ) appears to play a vital role in both adipocyte differentiation and insulin action, underscoring the important interactions between these two phenomena (105,106,107). A direct link between insulin sensitivity and PPARγ action in humans was strongly suggested by the discovery of a small number of individuals with heterozygous loss-of-function mutations within the ligand-binding domain of the PPARγ receptor (108,109,110). These dominant-negative and other mutations result in the clinical features of severe insulin resistance described above and partial lipodystrophy of the limbs and buttocks, sparing the face and central abdominal adipose depots (111). The associated lipodystrophy appears to result from the inability of subcutaneous abdominal adipose tissue to trap and store FFA postprandially (112). Although thiazolidinediones are of limited benefit in this syndrome, the tyrosine-based receptor agonist farglitazar corrected defects in ligand binding and restored transcriptional function in cells from affected individuals and may offer a therapeutic approach to improving insulin action in these patients.
By contrast, a missense mutation at codon 115 (Pro115Gln) was associated with severe obesity in four unrelated German subjects (113). Overexpression of the mutant receptor in murine fibroblasts led to accelerated differentiation of the cells into adipocytes and greater cellular accumulation of triglyceride when compared with the wild-type receptor. Although it might be expected that this constitutively active form of PPARγ might cause heightened insulin sensitivity, interpretation of the lower insulin levels in these subjects relative to obese controls was complicated by the fact that three of the four subjects had type 2 diabetes.
Of potentially more general relevance to type 2 diabetes, a common polymorphism in the PPAR γ gene may confer protection from type 2 diabetes (114,115). A Pro12Ala polymorphism in the PPARγ gene was associated with improved insulin sensitivity among more than 1,000 middle-aged and elderly Finns (114).This mutation occurs in the region of the PPARγ gene that encodes a protein segment unique to the PPARγ2 isoform and is within the domain of PPARγ that enhances ligand-independent activation (116). Subsequently, an analysis of more than 3,000 Scandinavian individuals in a study using a multilayered design revealed that this common polymorphism was associated with a small but significant reduction in risk of type 2 diabetes (relative risk 0.78) relative to the prevalent proline form (115). In contrast with an earlier report (114), a recent meta-analysis demonstrated an increase in body mass index (BMI) among obese (but not lean) Ala12 homozygotes (117). The disparate effects of the PPARγ gene Pro12Ala polymorphism on BMI in obese and lean individuals suggest that the impact of this genetic variant can be
modified by other environmental and/or genetic factors. In general, PPARγ expression is increased in the adipose tissue of obese subjects, but a low-calorie diet can downregulate its expression (118). It has been shown that when the dietary ratio of polyunsaturated fat to saturated fat is low, the BMI in Ala12 carriers is greater than that in Pro12 homozygotes, but when the ratio is high, the opposite is seen (119). This gene-nutrient interaction may explain, in part, the disparate effects of the Ala12 variant on BMI in obese and lean subjects.
modified by other environmental and/or genetic factors. In general, PPARγ expression is increased in the adipose tissue of obese subjects, but a low-calorie diet can downregulate its expression (118). It has been shown that when the dietary ratio of polyunsaturated fat to saturated fat is low, the BMI in Ala12 carriers is greater than that in Pro12 homozygotes, but when the ratio is high, the opposite is seen (119). This gene-nutrient interaction may explain, in part, the disparate effects of the Ala12 variant on BMI in obese and lean subjects.
LIPODYSTROPHY
This diverse group of disorders is characterized by severe insulin resistance and partial or complete absence of adipose tissue. The study of these rare genetic variants is important in gaining understanding of the relationship between body-fat distribution and insulin resistance. Following earlier descriptions of affected individuals, Dunnigan (120) first described a family with the syndrome of familial partial lipodystrophy (FPLD) in 1974. Patients with FPLD appear normal at birth but lose subcutaneous fat from their extremities and the gluteal region after puberty. This results in prominent, well-defined musculature in these areas, while fat deposition within the face and neck, axillae, back, labia majora, and abdominal cavity is markedly increased (121). Magnetic resonance imaging studies of these individuals have revealed a complete lack of subcutaneous fat in affected areas, with preservation of intermuscular, intraabdominal, intrathoracic, and bone-marrow fat and excessive accumulation of intramuscular fat (122). Recent genetic studies in families with FPLD have identified several mutations in the LMNA gene, which encodes lamin A and C (123,124). These proteins perform important structural functions in the nuclear membranes of adipocytes and myocytes and participate in DNA replication in these terminally differentiated cells (125). The reason for the regional distribution of the lipodystrophy remains to be determined.
By contrast, congenital generalized lipodystrophy is characterized by a total lack of adipose tissue and by insulin resistance from birth or infancy. The clinical features of insulin resistance (described above) are accompanied by an anabolic syndrome with muscular hypertrophy, hepatomegaly, and hypertrophic cardiomyopathy, the last being frequently lethal by early adulthood (126). This striking clinical syndrome is associated with mutations of two distinct genes on different chromosomes: the gene (AGPAT2) encoding 1-acylglycerol-3-phosphate- O -acyltransferase and a gene homologous to the murine guanine nucleotide-binding protein (G protein), BSCL2. Because the AGPAT2 enzyme catalyzes the acylation of lysophosphatidic acid to form phosphatidic acid, a key intermediate in the biosynthesis of triacylglycerol and glycerophospholipids, mutations in AGPAT2 probably cause congenital generalized lipo-dystrophy by inhibiting triacylglycerol synthesis and storage in adipocytes (127). The gene BSCL2 is most highly expressed in brain and testis and encodes a protein (seipin) of unknown function. Most of the variants are null mutations and probably result in a severe disruption of the protein (128). Potential mechanisms whereby lipodystrophy may cause insulin resistance are discussed below.
The various monogenic syndromes of insulin resistance described above reveal that there is considerable heterogeneity of phenotype among those affected with the identified mutations. Because this heterogeneity does not necessarily appear to be explained by gene penetrance or degree of defect in insulin signaling, it suggests that other factors (genetic or environmental or both) may be important in determining phenotype, even in these extreme genetic variants.
Commonly Occurring Polymorphisms
In addition to the extreme forms of insulin resistance associated with the rare genetic mutations described above, analysis of more commonly occurring polymorphisms of genes with relevance to insulin action has revealed the association of several such polymorphisms with increased incidence of type 2 diabetes.
INSULIN-SIGNALING PATHWAY
In a Caucasian population, individuals homozygous for a common codon 326Met>Ile variant of the p85 subunit of PI 3-kinase demonstrated reduced insulin sensitivity, although no increased incidence of type 2 diabetes (129). Several IRS-1 polymorphisms are more common among individuals with type 2 diabetes than among nondiabetic controls and have been studied extensively in many populations (130). Obese carriers of the G972R polymorphism in a Caucasian population demonstrated decreased insulin sensitivity and increased prevalence of metabolic cardiovascular risk factors (130). Transfection of this common IRS-1 variant into myeloid progenitor cells stably overexpressing the insulin receptor resulted in decreased binding of the p85 regulatory subunit of PI 3-kinase to IRS-1 and decreased IRS-1–associated PI 3-kinase activity (131). In a Japanese population, the combined prevalence of several IRS-1 polymorphisms was threefold greater among patients with type 2 diabetes, and both diabetic and nondiabetic carriers of the various polymorphisms demonstrated reduced insulin sensitivity (132).
Several genetic studies have examined whether polymorphisms in the locus for adiponectin, 3q27, could affect the circulating levels of adiponectin and whether these polymorphisms were associated with increased risk for the development of type 2 diabetes. Evidence for the effects of this adipose-specific circulating protein on insulin action will be discussed below. The results of one study showed evidence of linkage with the metabolic syndrome (133), whereas another showed evidence of a type 2 diabetes susceptibility locus at 3q27 in a French population with early-onset diabetes (134). Polymorphisms within the adiponectin locus were also linked with increased risk for type 2 diabetes in a Japanese cohort (135).
DIGENIC
There are a few examples of potential interactions between separate genes in conferring increased risk of insulin resistance (136). The combined effect of PC-1 (K121Q) and PPARγ2 (P12A) polymorphisms results in significant increases in BMI and insulin levels and impairments in both insulin sensitivity and insulin secretion. Additionally, a family has been described in which five individuals with severe insulin resistance (but no unaffected family members) were doubly heterozygous for frameshift/premature stop mutations in two unlinked genes, PPARG and PPP1R3A. These genes encode PPARγ and protein phosphatase 1 regulatory subunit 3, the muscle-specific regulatory subunit of protein phosphatase 1.
TRANSGENIC MOUSE MODELS OF HUMAN POLYGENIC BASIS OF DIABETES INHERITANCE
Given the complex genetic interactions that likely contribute to human diabetes, insulin receptor (Ir) heterozygous mutants have been used to mimic genetic interactions leading to type 2 diabetes. A first step in this direction was the development of a polygenic model of insulin-resistant diabetes by generating mice with combined heterozygous Ir and Irs1 mutations. Whereas Irs1 heterozygotes are normal and only ∼5% of Ir+/- heterozygotes develop diabetes, mice doubly heterozygous for both Ir- and Irs1-null alleles (Ir/Irs1+/-) develop severe
hyperinsulinemia and hyperplasia of pancreatic β-cells. Nearly half of these mice ultimately become frankly hyperglycemic (137), similar to the increased risk of diabetes in first-degree relatives of patients with type 2 diabetes (138). These characteristics of the Ir/Irs1+/- mouse are consistent with an oligogenic mode of inheritance of type 2 diabetes, in which two subclinical defects of gene function could account for virtually the entire genetic susceptibility to the disease. Even further increases in the incidence of diabetes occurred when these mice were crossed with mice doubly heterozygous for Ir and IRS-2 (Ir/Irs2+/-). Diabetes developed in 40% of the tripleheterozygotes (139), providing a useful experimental model for the complex genetic interactions that are likely to contribute to the inheritance of type 2 diabetes. Thus, even such a major predisposing allele as the null insulin receptor mutation has a modest effect alone but can contribute importantly to a predisposing background. This is confirmed by the widely variable prevalence of diabetes (<2% to 85%) observed when the double-heterozygous knockout mice are bred onto different genetic backgrounds (140).
hyperinsulinemia and hyperplasia of pancreatic β-cells. Nearly half of these mice ultimately become frankly hyperglycemic (137), similar to the increased risk of diabetes in first-degree relatives of patients with type 2 diabetes (138). These characteristics of the Ir/Irs1+/- mouse are consistent with an oligogenic mode of inheritance of type 2 diabetes, in which two subclinical defects of gene function could account for virtually the entire genetic susceptibility to the disease. Even further increases in the incidence of diabetes occurred when these mice were crossed with mice doubly heterozygous for Ir and IRS-2 (Ir/Irs2+/-). Diabetes developed in 40% of the tripleheterozygotes (139), providing a useful experimental model for the complex genetic interactions that are likely to contribute to the inheritance of type 2 diabetes. Thus, even such a major predisposing allele as the null insulin receptor mutation has a modest effect alone but can contribute importantly to a predisposing background. This is confirmed by the widely variable prevalence of diabetes (<2% to 85%) observed when the double-heterozygous knockout mice are bred onto different genetic backgrounds (140).
“THRIFTY GENOTYPE” VERSUS “THRIFTY PHENOTYPE”
These concepts highlight interesting aspects of gene-environment interactions. The high prevalence of insulin resistance and type 2 diabetes in certain population groups is consistent with effects of nutrient availability on gene selection over a long time. Because food supply was neither predictable nor consistent throughout most of the history of Homo sapiens, it is likely that ancestral hunter-gatherers experienced alternating cycles of feast and famine (141). It is postulated that the evolutionary pressures in the late Paleolithic era favored the selection of “thrifty genes” for efficient intake and utilization of fuel stores (142), thereby promoting storage of fuel for periods of impending famine. It does not appear that the genome has changed appreciably over the past 10,000 years, and certainly not with the explosion in the prevalence of type 2 diabetes during the past 40 to 100 years (143). Indeed, genes that favored survival in the late Paleolithic era would be maladaptive in the face of the current combination of continuous food abundance and physical inactivity. The complexity of gene-gene and gene-environment interactions discussed above suggests that the thrifty genotype is polygenic and includes genes that would impact an individual’s response to environmental factors.
There is also considerable epidemiologic evidence suggesting that nutrient availability may condition the patterns of an individual’s gene expression at early developmental stages. Indeed, intrauterine growth retardation is strongly linked with subsequent development of obesity or insulin resistance or both in later life (144). Many animal models have been generated to explore possible mechanisms for this association (145). In particular, maternal protein restriction has been shown to be associated with many metabolic defects in adult offspring, including resistance to the effects of insulin on peripheral glucose uptake (146) and hepatic glucose production (147), as well as defective insulin signaling in adipocytes (148) and hypertension (149). As opposed to the evolutionary pressures described above, the effects of fetal deprivation have been termed the “thrifty phenotype” (150). This suggests that the metabolism of the liver, muscle, and adipose tissue may be programmed by maternal nutrition during gestation and lactation and that an individual programmed to survive on minimal nutrients in utero may suffer adverse consequences in adult life in response to plentiful nutrition. Early growth restriction combined with subsequent supranormal nutrition in these experimental models results in the clinical features of the insulin resistance syndrome.
IMPACT OF OBESITY ON INSULIN ACTION
The quantity of body fat varies widely in mammals, ranging from 2% to 50% of body mass (151). This large variation in fat mass is apparently determined both by an individual’s genetic background (152) and by environmental factors, including diet and physical activity (153). Excess body fat, or obesity, is an important factor in the pathogenesis of insulin resistance and substantially increases the risk of type 2 diabetes.
The Obesity Epidemic
In the United States of America, successive cross-sectional nationally representative surveys such as the National Health and Nutrition Examination Surveys (NHANES I: 1971–1974; NHANES II: 1976–1980; NHANES III: 1988–1994) permit the study of trends in weight over time. Disturbingly, the prevalence of obesity (BMI ≤ 30.0) showed a large increase from NHANES II to NHANES III: NHANES I, 14.1%; NHANES II, 14.5%; and NHANES III, 22.5% (154). In NHANES III, the crude prevalence of overweight and obesity (BMI > 25.0) among adults was 59.4% for men, 50.7% for women, and 54.9% overall. During this time of rising prevalence in obesity, the levels of physical activity have decreased and important shifts in diet have occurred (155), including increased consumption of sugars and energy-dense foods (156). Average daily caloric intake by people in the United States aged 2 years and older increased by 194 kcal/day from 1977 to 1996, with the largest increases in caloric intake among adolescents and young adults (157).
This highly disturbing trend is not limited to the United States or even to developed countries (158). Analyses of economic and food availability data for 1962 through 1994 have revealed a major shift in the structure of the global diet, with an uncoupling of the classic relationship between incomes and fat intakes (159). Global availability of cheap vegetable oils and fats has resulted in greatly increased fat consumption among people in low-income nations, including many countries in Asia, Latin America, North Africa, the Middle East and the urban areas of sub-Saharan Africa. Consequently, obesity is becoming a significant health issue even in countries with lower levels of gross national product and is aggravated by high urbanization rates (160). As expected, dramatic increases in the consumption of low-cost fat and simple carbohydrate calories have resulted in steep increases in incidence of overweight and obesity even among the poorest individuals in these countries (161).
The following section will highlight the metabolic effects of obesity and putative mechanisms whereby obesity contributes to insulin resistance. The impact of increased nutrients will be examined separately below, in light of substantial evidence that both factors impact insulin action.
Adipose Tissue as an Endocrine Organ
Besides being the body’s principal site for energy storage, white adipose tissue influences whole-body insulin action both through release of FFAs and by secretion of adipose-derived proteins. The latter include both proinflammatory peptides, discussed below, and several newly identified hormones that appear to have dramatic effects on glucose metabolism and insulin action (162,163).
ADIPONECTIN
Adiponectin (also known as Acrp30, AdipoQ, and GBP28) is a 30-kDa adipose-specific secretory protein that appears to enhance insulin sensitivity (164,165). Decreased circulating
levels of this hormone accompany obesity and insulin resistance in both humans and animal models (166–168). Indeed, adiponectin levels were negatively correlated with hyperinsulinemia and degree of insulin resistance in both Pima Indian and Japanese groups (168,169). As described above, genetic polymorphisms of the adiponectin gene are associated with increased incidence of insulin resistance and diabetes. Although there do not appear to be acute effects of nutrient excess or deprivation on adiponectin levels, chronic caloric restriction (which enhances insulin action) increases adiponectin levels (170,171). Furthermore, high baseline plasma levels of adiponectin are associated with a substantially reduced risk of development of type 2 diabetes (172). The experimental models discussed below suggest that low plasma levels of adiponectin contribute to the pathogenesis of insulin resistance and type 2 diabetes.
levels of this hormone accompany obesity and insulin resistance in both humans and animal models (166–168). Indeed, adiponectin levels were negatively correlated with hyperinsulinemia and degree of insulin resistance in both Pima Indian and Japanese groups (168,169). As described above, genetic polymorphisms of the adiponectin gene are associated with increased incidence of insulin resistance and diabetes. Although there do not appear to be acute effects of nutrient excess or deprivation on adiponectin levels, chronic caloric restriction (which enhances insulin action) increases adiponectin levels (170,171). Furthermore, high baseline plasma levels of adiponectin are associated with a substantially reduced risk of development of type 2 diabetes (172). The experimental models discussed below suggest that low plasma levels of adiponectin contribute to the pathogenesis of insulin resistance and type 2 diabetes.
Pharmacologic treatment with adiponectin in rodents increases hepatic insulin sensitivity, with decreased glucose production. It was first noted that a single injection of purified recombinant adiponectin caused a transient decrease in basal glucose levels in various mouse models, with temporary correction of hyperglycemia without affecting insulin levels in ob/ob, nonobese diabetic, and streptozotocin-treated mice (173). Additionally, adiponectin increased the ability of subphysiologic levels of insulin to suppress glucose production in isolated hepatocytes (173). Infusion of adiponectin in conscious mice resulted in improved hepatic insulin action measured during euglycemic hyperinsulinemic clamp studies, with enhanced suppression of glucose production (174). Adiponectin stimulated phosphorylation and activation of the 5′AMP-activated protein kinase (AMPK) in skeletal muscle and in liver, together with stimulation of fatty acid oxidation and glucose uptake in myocytes, reduced gluconeogenesis in the liver, and reduction of glucose levels in vivo (175). Blocking AMPK activation inhibited all of these effects, suggesting that stimulation of glucose utilization and fatty acid oxidation by adiponectin occurs through activation of AMPK.
The phenotypes of adiponectin-deficient and transgenic adiponectin-overproducing animal models further substantiate an important role for adiponectin in mediating insulin action. Using a dominant mutation in the collagenous domain of adiponectin to generate a transgenic mouse, Combs et al (176). demonstrated that approximately threefold elevations in circulating adiponectin levels were associated with enhanced insulin-mediated suppression of endogenous glucose production and increased phosphorylation of AMPK in liver. Conversely, mouse models with a disruption of the adiponectin locus display moderate insulin resistance and impaired FFA clearance on a normal chow diet and an earlier onset of diet-induced insulin resistance (177,178).
It has recently been reported that adiponectin circulates in two discrete forms: as a relatively low-molecular-weight (LMW) hexamer and as a high-molecular-weight (HMW) multimeric structure (179). The ratio of these two oligomeric forms (HMW/ LMW) has recently been shown to correlate more tightly with insulin action than do total circulating adiponectin levels (180). Furthermore, although pharmacologic activation of PPARγ with thiazolidinediones increases adiponectin levels, the most striking effect is a selective proportional increase in the HMW form with close association with improved hepatic insulin action (180). This suggests that the HMW form is the active circulating form of adiponectin, and that its favorable effects on glucose metabolism are predominantly at the level of the liver.
LEPTIN
Leptin is a 16-kDa protein secreted from adipose tissue, the product of the defective obesity gene identified by positional cloning in the obese, hyperinsulinemic ob/ob mouse (181). Circulating leptin concentrations in humans correlate closely with fasting insulin concentrations and the percentage of body fat, making leptin a marker of obesity and the insulin resistance syndrome. Leptin infusion acutely increases glucose uptake under euglycemic, hyperinsulinemic conditions in rats (182). Indeed, acute leptin administration enhances the ability of insulin to inhibit hepatic glucose production but does not affect peripheral insulin action (183). By contrast, chronic leptin administration induces metabolically favorable changes in body composition that include decreased visceral adiposity and reduced muscle accumulation of triglyceride (184). These leptin-induced changes in body composition are associated with improved sensitivity to the metabolic effects of insulin on skeletal muscle (184).
Recent findings suggest that these favorable effects on body composition and lipid storage may be due to leptin-induced stimulation of β-oxidation via activation of AMPK in muscle and liver (185,186). When activated, AMPK decreases adenosine triphosphate (ATP)-consuming anabolic pathways such as glucose-regulated transcription, protein synthesis, cholesterol synthesis, and fatty acid and triglyceride synthesis and increases ATP-producing catabolic pathways such as increased glucose transport, β-oxidation, glycolysis, and mitochondrial biogenesis. Other potential mechanisms whereby leptin could influence insulin action include its regulation of immune function and hormone secretion (187). Profound leptin deficiency in ob/ob mice is accompanied by impaired T-cell immunity (188). Furthermore, the immunosuppression associated with acute starvation is reversed when exogenous leptin is administered (189). Leptin also alters the regulation of hormones in the hypothalamus-pituitary-adrenal axis and affects growth hormone, prolactin, and a number of other anterior pituitary hormones (190), many of whose effects on insulin action are discussed above. A discussion of mechanisms whereby leptin deficiency contributes to hyperinsulinemia and insulin resistance in lipodystrophy is provided below. Thus, defective leptin action may contribute to insulin resistance in the metabolic syndrome.
It has been postulated that leptin resistance can develop in the face of high circulating levels of the hormone. This is supported by the fact that leptin levels are increased in most mouse models of insulin resistance associated with obesity. Furthermore, diet-induced obese mice are resistant to the effects of leptin administration (191). Indeed, resistance to the central anorectic effects of leptin develops rapidly in the face of nutrient excess (192). However, leptin was effective in increasing energy expenditure in obese individuals after a sustained weight reduction of ∼10% of their previous body weight (193). This suggests that it may be possible to regain leptin sensitivity with weight loss and/or nutrient restriction, which may also have favorable implications for insulin action.
RESISTIN
Resistin is a 10-kDa adipose tissue-specific hormone recently identified in a screen for transcripts upregulated during adipogenesis and downregulated by treatment with a PPARγ agonist (194). Whereas injection of resistin into wild-type mice impaired glucose tolerance and insulin action, neutralizing antibodies improved insulin action in obese diabetic mice (194). When resistin was infused at near-physiologic levels in normal rats, lower rates of glucose infusion were necessary to maintain basal glucose levels during a hyperinsulinemic clamp study (195). The insulin resistance caused by resistin infusion was wholly accounted for by an increase in the rate of glucose production, suggesting that resistin has rapid inhibitory effects on hepatic rather than peripheral insulin sensitivity.
Despite this compelling evidence for acute effects of resistin on in vivo insulin action, there remains considerable uncertainty about the significance of resistin to the long-term regulation of insulin action in animals and humans. Indeed, resistin mRNA appears to be downregulated in most mouse models of insulin resistance (196). Additionally, resistin expression was very low in isolated adipocytes from human adipose tissue explants (197) and was actually higher in monocytes (198) and other nonadipocyte cells of adipose tissue (199) than in adipocytes. Furthermore, although some studies suggest a link between resistin levels and BMI or insulin sensitivity in humans (200,201,202), others have failed to reveal such a connection (203,204,205). However, it is possible that serum levels do not correlate with tissue mRNA or protein levels, a phenomenon observed for other adipokines under certain conditions (206). Given the challenges of reliably measuring plasma levels of resistin, most published papers on this topic report only tissue resistin transcript levels, and therefore more studies will be necessary to determine the clinical relevance of resistin in obesity and in the development of insulin resistance.
Obesity and Inflammation
There is growing evidence that the relationship between inflammation and insulin resistance is not merely correlative but actually causative (207). Epidemiologic data from several large studies have demonstrated a link between insulin resistance and systemic inflammation in both type 2 diabetes and nondiabetic populations (208,209,210). Adipose tissue produces many proinflammatory molecules, including TNF-α, IL-6, transforming growth factor-β, C-reactive protein, and monocyte chemotactic (chemoattractant) protein-1 (MCP-1), and circulating levels of these “adipokines” are increased in obesity (211,212,213,214,215,216).
Adipose-derived proinflammatory molecules are believed to induce systemic insulin resistance and to contribute to the pathogenesis of many metabolic complications of obesity, including type 2 diabetes and atherosclerosis (210). Although definitive human evidence is still accumulating, substantial experimental evidence links proinflammatory molecules with insulin resistance. The proinflammatory cytokine TNF-α has been demonstrated to mediate insulin resistance as a result of obesity in several rodent models (217). Expression of TNF-α was increased in white adipose tissue from different rodent models of obesity and diabetes (217), with elevations in TNF-α protein both locally and systemically. Neutralization of TNF-α in insulin-resistant rats caused a significant increase in the peripheral uptake of glucose in response to insulin (217,218). Furthermore, mice lacking either the TNF-α ligand or one of its receptors were partially protected from obesity-induced insulin resistance (219,220). TNF-α directly decreases insulin sensitivity and increases lipolysis in adipocytes (221,222). Expression of TNF-α is increased in white adipose tissue in obese and insulin-resistant states (223). Multiple mechanisms have been suggested to account for these metabolic effects of TNF-α, including direct effects on insulin signaling, downregulation of genes that are required for normal insulin action, induction of elevated FFA levels via stimulation of lipolysis, and negative regulation of PPARα (224).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


