Insulin-like Growth Factors
Karen C. McCowen
Robert J. Smith
Insulin-like growth factors (IGFs) belong to a family of peptide hormones with structural and functional similarities to insulin. Initially identified on the basis of their insulin-like glucoregulatory effects, IGFs were given the name nonsuppressible insulin-like activity, because hypoglycemia failed to lower their levels in the circulation. Nonsuppressible insulin-like activity was recognized subsequently to consist of various forms of two related proteins, which now are designated insulin-like growth factor-I (IGF-I) and IGF-II. This chapter will focus primarily on IGF-I, a peptide hormone of 7,600 daltons, which is the major mediator of growth-promoting actions of growth hormone. It is present in the circulation at significant levels throughout postnatal life and has glucoregulatory and mitogenic properties that are analogous to those of insulin. IGF-II is related in structure to IGF-I but the product of a separate gene. IGF-II is believed to have important roles in fetal growth and differentiation plus effects on postnatal growth in some tissues. The production of IGF-II by extrapancreatic neoplasms of various types represents an uncommon but well-established cause of tumor-associated hypoglycemia (see Chapter 69 for further discussion).
Unlike insulin, which is derived from a single cell type (pancreatic β-cells) and functions entirely as an endocrine hormone, IGF-I is produced in many different tissues and often acts in a paracrine or autocrine manner. Thus, appreciation of IGF-I function in vivo is more difficult than that of a classical endocrine hormone. Insulin and IGFs have common ancestral origins; IGF-I is postulated to have evolved more than 300 million years ago from a single primordial insulin-IGF gene through intron rearrangement followed by gene duplication (1). IGF-I is secreted intact, as it is produced (i.e., not stored intracellularly), and is composed of four domains designated A through D (Figure 10.1). The A- and B-domains are analogous to the A- and B-chains of the insulin molecule. The C-domain is retained in mature IGF-I and IGF-II, whereas the equivalent region is excised during processing of proinsulin to insulin. The D-domain also is present in IGF-I and IGF-II but absent from insulin. The potential for cross-reactivity of IGF-I with insulin receptors, as well as insulin binding to IGF-I receptors, may contribute to complications associated with insulin resistance and diabetes. Despite their substantial homology and capacity for activation of similar cellular signaling pathways, IGF-I and insulin have distinct biologic effects on target tissues. However, IGF-I infusions can occasionally be used to decrease blood glucose in cases of extreme insulin resistance, with IGF-I acting either through its own receptor or through the insulin receptor. The major role of IGF-I in vivo is most likely its involvement in cell division, growth, and proliferation. IGF-I has been implicated in proliferative aspects of certain complications of diabetes, such as retinopathy and nephropathy, although further research is needed to establish the pathologic importance of IGF-I in diabetes complications, and major therapeutic implications have not been realized.
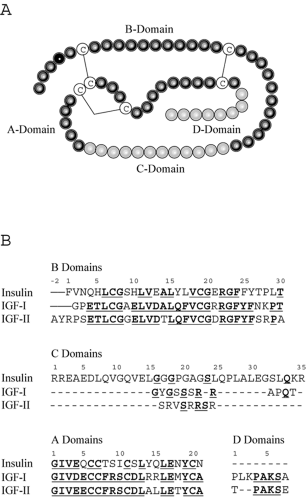 Figure 10.1. A: Domain structure of the insulin-like growth factor-I (IGF-I) protein. The single polypeptide chain extends from the amino-terminal B-domain through the C-domain and A-domain to the carboxy-terminal D-domain. The amino acids labeled C represent cysteines, which are linked by disulfide bonds as shown. B: Comparative amino acid sequences of human insulin, IGF-I, and IGF-II. Amino acids that are identical in two or more of these hormones are underlined in bold. |
INSULIN-LIKE GROWTH FACTOR-I STRUCTURE AND REGULATION
Circulating IGF-I is substantially derived from liver parenchymal cells, which are stimulated to synthesize and secrete the hormone in response to growth hormone (GH) acting through
hepatic GH receptors. However, IGF-I can also be synthesized locally in a multitude of different tissues under the influence of GH and, to some extent, independently of GH. As shown in Figure 10.1B, IGF-I is homologous to insulin and, even more so, to proinsulin, which includes the C-domain region. Major structural differences are found in the first 16 amino acids of the B-chain, such that IGF-I has considerable affinity for a family of binding proteins that modulate both its circulating half-life and its accessibility to tissues.
hepatic GH receptors. However, IGF-I can also be synthesized locally in a multitude of different tissues under the influence of GH and, to some extent, independently of GH. As shown in Figure 10.1B, IGF-I is homologous to insulin and, even more so, to proinsulin, which includes the C-domain region. Major structural differences are found in the first 16 amino acids of the B-chain, such that IGF-I has considerable affinity for a family of binding proteins that modulate both its circulating half-life and its accessibility to tissues.
Although GH is an important regulator of IGF-I synthesis and secretion, nutritional status and the presence of hyperglycemia also have important influences on concentrations of IGF-I. GH is secreted in a pulsatile and diurnal variable manner from the anterior pituitary under the influence of both positive and negative hypothalamic peptides. GH concentrations peak in the early morning, and because GH acts as a promoter of insulin resistance, rising levels of GH may contribute to early-morning insulin resistance, the “dawn phenomenon” that occurs commonly in diabetes mellitus. The GH receptor is dimerized upon activation by GH and transmits downstream signals by effecting tyrosine phosphorylation of proteins from both the JAK and STAT families, as well as by activating other signaling intermediates, ultimately leading to changes in ex-pression of genes, including those encoding IGF-I and IGF-binding protein-3 (Fig. 10.2).
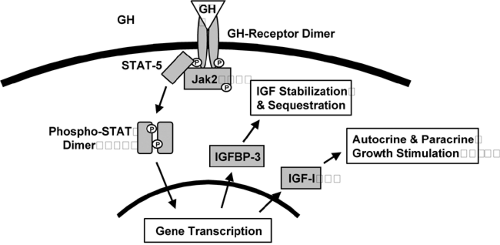 Figure 10.2. Growth hormone (GH) signaling pathways involving initial cytoplasmic events and consequent changes in nuclear gene transcription. IGF, insulin-like growth factor; IGFBP-3, insulin-like growth factor-binding protein-3; P, phosphotyrosine. |
Malnutrition, either protein malnutrition or generalized protein-calorie malnutrition (marasmus), also affects IGF-I gene transcription. GH resistance has been reported in malnutrition, with elevated concentrations of GH (2) but low IGF-I mRNA in liver tissue (3,4) and low serum IGF-I (5,6). Fasting has been shown to reduce circulating IGF-I in healthy persons, in association with elevated integrated GH concentrations (7). In rats, GH-binding sites on hepatocytes were reduced following a fast, and GH binding increased in concert with IGF-I plasma concentrations during refeeding (8). In contrast to the effects of overall nutrient restriction, isolated dietary protein depletion results in decreased IGF-I levels in the absence of a change in liver GH-binding capacity, suggesting that postreceptor mechanisms are responsible for the decline in IGF-I in this situation (9,10,11). Nutritional repletion increases levels of both IGF-I mRNA and protein, and increasing IGF-I concentrations have been shown to predict a return to normal nitrogen balance in ill patients starting nutritional support (12,13). Interestingly, the provision of both energy and protein is required to restore
IGF-I concentrations. In malnourished laboratory rats, refeeding a eucaloric but low-protein diet was adequate to restore IGF-I, whereas providing protein at 1 g/kg in the face of marked energy restriction was insufficient (14,15). When protein intake is limiting, essential amino acids are more effective at raising IGF-I than is a nonspecific mixture of amino acids (16), although in nutritional support of hospitalized patients, use of essential amino acids has not been associated with improvement in clinical endpoints. As a source of energy, dietary fat appeared to be less effective than carbohydrate in raising IGF-I concentrations and in improving nitrogen balance in a study examining responses to exogenous GH administration in the presence of limited energy intake (15). Hypercaloric nutrition in healthy volunteers has been associated with slight increases in circulating IGF-I, but the effect of overnutrition appears to be much less marked than that of undernutrition (17).
IGF-I concentrations. In malnourished laboratory rats, refeeding a eucaloric but low-protein diet was adequate to restore IGF-I, whereas providing protein at 1 g/kg in the face of marked energy restriction was insufficient (14,15). When protein intake is limiting, essential amino acids are more effective at raising IGF-I than is a nonspecific mixture of amino acids (16), although in nutritional support of hospitalized patients, use of essential amino acids has not been associated with improvement in clinical endpoints. As a source of energy, dietary fat appeared to be less effective than carbohydrate in raising IGF-I concentrations and in improving nitrogen balance in a study examining responses to exogenous GH administration in the presence of limited energy intake (15). Hypercaloric nutrition in healthy volunteers has been associated with slight increases in circulating IGF-I, but the effect of overnutrition appears to be much less marked than that of undernutrition (17).
Uncontrolled diabetes is associated with low serum IGF-I concentrations, although whether diabetes has an effect independent of concomitant negative nutritional status has not been determined (18). Insulin-deficient diabetic rats have low IGF-I concentrations, which are restored at least partially with exogenous insulin therapy (19,20,21). Although nutritional factors confound the relationship between the two hormones, the finding that exposure to insulin in vitro increases the number of GH receptors on hepatocytes supports a direct link between insulin status and IGF-I levels (22). Protein restriction in diabetic rats prevents the restoration of IGF-I concentrations following insulin treatment, in contrast to a normalization of IGF-I in insulin-treated diabetic rats given adequate dietary protein (23).
Circulating IGF-I is bound with high affinity to a number of IGF-binding proteins (IGFBPs). Six binding proteins in the range of 20 to 30 kDa have been cloned and sequenced. The bulk (∼90%) of measurable IGF-I is bound to IGF-binding protein-3 (IGFBP-3), with only a small fraction (<5%) of total IGF-I being free. IGFBP-3 serves as a reservoir of IGF-I, which retains IGF-I in the circulation, prevents rapid fluctuations in plasma concentrations, and probably functions to prolong the circulating half-life of IGF-I. IGFBP-3 is generally regulated in parallel with IGF-I, with nutrition and GH being major stimulators of IGFBP-3 concentrations. IGFBP-1, forming a smaller complex with IGF-I, can cross capillary endothelium and may participate in the delivery of IGF-I to the cell surface, where IGF-I receptors are accessible. Food intake, in association with rising plasma concentrations of insulin, lowers IGFBP-1 concentrations abruptly, perhaps by inducing its movement out of the vascular lumen (24). Both IGFBP-1 and IGFBP-2 concentrations increase with fasting (25) and, in the presence of diabetes, show opposite regulation from IGFBP-3 and IGF-I (26,27). Assessment of the kinetics of IGF-I action is difficult, because the functions and importance of the IGFBPs have not been fully defined. Synthetic analogues of IGF-I, altered so that binding to IGFBPs is prevented, are more potent than IGF-I when injected into experimental animals (28,29). This suggests that inhibition of IGF-I action is a major function of the binding proteins, although the binding proteins also may have some actions independent of the IGFs (30).
INSULIN-LIKE GROWTH FACTOR-I RECEPTOR
The IGF-I receptor (also designated type I IGF receptor) is pres-ent in multiple body tissues, with the notable exceptions of liver parenchymal cells and adipocytes, in which the levels are very low. IGF-I receptor mRNA is most abundant during embryonic life, with significant but much lower levels during adulthood. The functional receptor is a heterotetrameric transmembrane protein containing two types of subunits joined by disulfide bonds in a B-A-A-B configuration (Fig. 10.3). This is closely analogous to the structure of the insulin receptor. The α-subunits (Mr 135 kDa) are entirely extracellular and contain the hormone-binding site; the β-subunits (95–105 kDa, depending on the tissue) span the plasma membrane, and each contains an intracellular tyrosine kinase domain. IGF-I and insulin recep-tors are encoded by separate genes, but they exhibit approximately
40% overall amino acid sequence identity, and there is phylogenetic evidence for their emergence from a single precursor receptor. Much higher sequence homology is present in key functional regions, such as the tyrosine kinase domain. In spite of extensive similarities, the critical regions involved in hormone binding establish clear functional differences for insulin and IGF-I receptors. IGF-I and insulin each binds with high affinity to its cognate receptor. There is a potential for binding cross-reactivity related to structural similarities of the ligands and receptors, although such binding generally requires concentrations of the opposing hormone above the normal physiologic range (Fig. 10.3). Thus, the IGF-I receptor binds insulin with much lower affinity than it binds IGF-I.
40% overall amino acid sequence identity, and there is phylogenetic evidence for their emergence from a single precursor receptor. Much higher sequence homology is present in key functional regions, such as the tyrosine kinase domain. In spite of extensive similarities, the critical regions involved in hormone binding establish clear functional differences for insulin and IGF-I receptors. IGF-I and insulin each binds with high affinity to its cognate receptor. There is a potential for binding cross-reactivity related to structural similarities of the ligands and receptors, although such binding generally requires concentrations of the opposing hormone above the normal physiologic range (Fig. 10.3). Thus, the IGF-I receptor binds insulin with much lower affinity than it binds IGF-I.
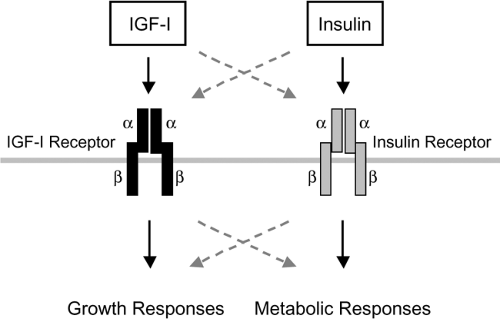 Figure 10.3. Homology in the structures of the insulin-like growth factor-I (IGF-I) and insulin receptors. Each hormone binds with greatest affinity to its own receptor but when present at high concentrations can bind to the other receptor. Both receptors can induce a full complement of cellular sig-naling responses. However, there is evidence for greater stimulation of cell-growth responses by the IGF-I receptor and greater stimulation of metabolic responses by the insulin receptor. |
Hormone-binding specificity depends on key cysteine residues and other structural features in the extracellular receptor α-subunits, whereas the transmembrane β-subunits mediate signal transduction predominantly via their intrinsic tyrosine kinase activity. Binding of IGF-I to its receptors is believed to result in conformational changes that lead to autophosphorylation in a “trans” reaction, in which the tyrosine kinase of one β-chain cross-phosphorylates specific tyrosine residues in the opposite β-chain within the receptor heterotetramer. This positions the active tyrosine kinase domains for the phosphorylation of cellular substrates, such as the insulin receptor substrate (IRS) proteins, which then initiate a cascade of downstream signaling events that result in hormone action (see Chapter 9 for further details on the signaling pathways of the insulin and IGF-I receptors).
INSULIN AND INSULIN-LIKE GROWTH FACTOR-I SPECIFICITY
Although insulin and IGF-I are structurally similar and, at high concentrations, can activate each other’s receptors, the receptors provide substantial signaling specificity. IGF-I is approximately 1,000 times less potent than insulin in displacing radiolabeled insulin from its insulin-receptor binding sites. Insulin, in turn, is approximately 500 times less potent than IGF-I in displacing radiolabeled IGF-I from its IGF-I—receptor binding sites. On a molar basis, injected IGF-I is only about 6% as potent as insulin in causing hypoglycemia in humans (31). IGF-I differs from insulin not only in being produced in a wide variety of tissues but in having markedly different diurnal circulating profiles, with an absence of clear peaks for IGF-I. In skeletal muscle biopsies from obese diabetic humans, binding of insulin to its receptors was markedly diminished, but binding of IGF-I was unchanged. IGF-I-stimulated tyrosine kinase activity examined ex vivo was also not different in tissue extracts from diabetic and control subjects (32). This suggests discrete downstream pathways with differential regulation of insulin and IGF-I receptors. In another study addressing the same issue, skeletal muscle from nondiabetic individuals demonstrated a twofold increase in glucose transport in response to IGF-I, whereas samples from diabetic patients showed no activation of glucose transport. It was observed, however, that binding of IGF-I to skeletal muscle was normal in the diabetic patients (33). This suggests that there may be changes specific to the insulin receptor in type 2 diabetes but that resistance to both insulin and IGF-I is at a common site downstream of the receptor tyrosine kinases.
Insulin signaling through its receptor activates phosphorylation cascades that lead to both metabolic and mitogenic cellular responses (Fig. 10.3), though the most important actions of insulin are thought to affect metabolic endpoints. Although the binding of IGF-I to its receptors can activate many of the same signaling molecules, IGF-I actions are thought to be primarily mitogenic. In cultured fibroblasts expressing similar numbers of insulin and IGF-I receptors, IGF-I activation of pathways leading to DNA synthesis was more marked, suggesting quantitative differences in the stimulation of different postreceptor pathways by IGF-I and insulin receptors (34). Studies on chimeric receptor proteins containing the receptor-binding portion of a neurotrophin receptor fused to the intracellular domains of either IGF-I or insulin receptors indicate that at least part of receptor signaling specificity lies within the intracellular domains (35). When activated by neurotrophin, the insulin-receptor chimera expressed in cultured fat cells was more effective in stimulating glucose transport and translocation of the GLUT4 glucose transporter. By contrast, when expressed in the same fat cell line, the IGF-I-receptor chimera more effectively activated mitogen-activated protein (MAP) kinase pathways,
which are believed to link most strongly to the stimulation of DNA synthesis and cell proliferation.
which are believed to link most strongly to the stimulation of DNA synthesis and cell proliferation.
Investigation of other signaling differences between insulin and IGF-I receptors that confer specificity on each pathway is critically important to our understanding of IGF-I action in vivo. Evidence continues to accumulate that differences between these two signaling pathways exist, although there also appears to be substantial overlap. The use of peptides containing four major phosphorylation sites of IRS-1 in kinase assays with each receptor showed identical specificity of insulin and IGF-I receptors for IRS-1 (36). Although the two hormones activate IRS-1 similarly (at least in vitro), a specific isoform of phosphoinositide 3-kinase (PI 3-K-C2α), a signaling enzyme activated by IRS-1, was stimulated to a greater extent by insulin receptors than by IGF-I receptors (37). The liver plasma membrane glycoprotein, pp120, also was found to be preferentially phosphorylated by insulin receptors in cultured fibroblasts transfected with insulin as compared with IGF-I receptors (38). By contrast, IGF-I receptors stimulated tyrosine phosphorylation of the signaling adapter Crk II, whereas insulin receptors were substantially less effective (39).
Specificity in IGF-I and insulin action may result from the distinct interactions of these two receptor proteins not only with signal-transducing molecules but also with signaling regulators. For example, Grb10 is a putative adapter protein that has the capacity to bind to IGF-I and insulin receptors through the interaction of motifs in its C-terminal region with phosphotyrosine residues in activated IGF-I and insulin receptors (40,41,42). Evidence has been presented for both negative and positive effects of Grb10 on IGF-I and insulin signaling. In studies on a specific isoform of Grb10 in mouse tissues (Grb10Δ), much stronger interaction was demonstrated with insulin receptors than with IGF-I receptors (43).
cDNA microarray technology has been used to examine the expression levels of genes in cultured fibroblasts overexpressing either insulin or IGF-I receptors (44). IGF-I activation of its receptors led to increased mRNA levels for 30 genes that were not increased as a consequence of insulin activation of insulin receptors. These were mostly genes known to be important in growth and differentiation. A total of nine genes were much more strongly stimulated by insulin receptors than by IGF-I receptors, none of which had previously been noted to be insulin responsive. The implications of this study are yet unclear because the observations were made only in cultured fibroblast cell lines. However, these data add further support to the concept that IGF-I and insulin receptors mediate distinct signaling responses in their target cells.
Insulin and IGF-I receptors are required for normal embryonic growth and development, but their roles early in development are different from those in postnatal life (45). IGF-I receptors mediate the effects of both IGF-I and IGF-II during embryonic development, whereas insulin receptors mediate effects of IGF-II but not those of insulin (46). Humans with homozygous or compound heterozygous mutations of the insulin receptor who present at birth with the leprechaunism syndrome (a severe form of insulin resistance) have severe intrauterine growth retardation and postprandial hyperglycemia, and die in infancy (47). In fibroblasts derived from a patient with this syndrome, IGF-I binding and IGF-I receptor β-subunit tyrosine phosphorylation were normal, as was IGF-I-; stimulated thymidine incorporation into DNA, suggesting the potential therapeutic usefulness of IGF-I (48). Clinical investigations in this patient demonstrated low circulating concentrations of IGF-I and IGFBP-3, which were resistant to GH therapy. Administration of recombinant human IGF-I maintained near-normal growth and glycohemoglobin for 6 years. Tonsillar hypertrophy was the only evident complication of IGF-I therapy, as previously has been observed following IGF-I treatment of patients with defects in the GH receptor (Laron dwarfism). Treatment of other patients with insulin-receptor mutations with IGF-I has yielded variable success, with evidence in some patients of combined resistance to both insulin and IGF-I (49). Since IGF-I and insulin receptors exist in part as hybrid constructs composed of one IGF-I and one insulin-receptor αβ-heterodimer, it is possible that some mutated forms of insulin half-receptor could exert a dominant negative effect on the IGF-I half-receptor and thus result in IGF-I resistance.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


