- Insulin signaling is accurately viewed as involving a matrix of interacting pathways, allowing for extensive modulation and divergence in signal transduction, rather than a linear cascade of sequential reactions.
- Diverse cell types commonly share the proximal steps in insulin signal transduction, including the insulin receptor, insulin receptor substrate molecules (IRS), phosphatidylinositide 3 kinase and Akt/protein kinase B.
- Insulin action is negatively modulated by multiple cellular mechanisms that impair tyrosine phosphorylation of insulin receptors and IRS; foremost among these are serine–threonine kinases and protein tyrosine phosphatases.
- The tissue-specific biologic effects of insulin are explained by effector systems which are uniquely expressed in differentiated target tissues such as the insulin-responsive glucose transport system in skeletal muscle, enzymatic systems mediating antilipolysis in adipose tissue and regulated gene expression leading to suppression of gluconeogenesis in liver.
- Insulin action is highly regulated by three pathways for nutrient sensing: the hexosamine biosynthetic signaling pathway; the mammalian target of rapamycin signaling pathway; and the AMP-activated protein kinase signaling pathway.
- Oxidative stress, inflammation and endoplasmic reticulum stress are associated with insulin resistance, obesity and metabolic syndrome, and impair insulin action via activation of serine–threonine protein kinases.
General aspects of insulin action
The peptide hormone insulin is exclusively synthesized in and secreted from pancreatic β-cells. Insulin exerts a broad spectrum of anabolic effects in multiple tissues. The regulation of whole body fuel homeostasis primarily involves insulin action in skeletal muscle, adipose tissue, and liver where insulin promotes uptake and storage of carbohydrate, fat, and amino acids, while at the same time antagonizing the catabolism of these fuel reserves. In skeletal muscle, insulin stimulates glucose transport and glucose storage as glycogen, as well as glycolysis and tricarboxylic acid cycle activity. Insulin lowers hepatic glucose output by inhibiting glycogenolysis and gluconeogenesis, and augments glycogen formation. In adipocytes, insulin promotes glucose uptake, glycerol synthesis, and triglyceride formation, while at the same time exerting an antilipolytic effect. During periods of fasting, a fall in circulating insulin combined with increased secretion of counter-regulatory hormones leads to breakdown of stored fuels and increased availability of metabolic substrates for cellular energy.
In this way, alterations in insulin levels in the fed and fasting states have a key role in fuel metabolism and maintain blood glucose levels within a narrowly defined range. Insulin diminishes protein catabolism and increases translation, and also enhances cell growth, differentiation, and survival as a consequence of mitogenic and anti-apoptotic processes. Thus, the action of insulin at the level of cells and tissues affects substrate flux and coordinates the function of multiple organs as whole organisms adapt to the nutritional environment.
In mediating its pleiotropic actions, insulin binds to cell surface receptors, activates multiple signal transduction networks, and engages effector systems responsible for specific biologic functions. Over the last decade it has become clear that the classic perspective of insulin signaling as a linear cascade of sequentially interacting signal transduction molecules is short-sighted. Rather, the promulgation of insulin action is more accurately viewed as alterations in a network of interactions involving a matrix of signal cascades that engage in cross-talk. The final biologic action represents the net synergism of the combined facilitative, inhibitory and complementary signaling pathways that interact with more terminal functional systems in cell biology. While there is relative commonality in signal transduction networks, differentiated insulin target cells express a variety of unique effector systems, which are primarily responsible for mediating the cell specific and organ-specific biologic functions of insulin. Effector systems include rate-limiting enzymes, enzymatic pathways, membrane transport systems, gene expression, processes regulating the cellular trafficking of proteins and vesicles, and systems governing the translation, post-translational modification and degradation of proteins. Certain aspects of linear insulin signal transduction are evolutionarily conserved; however, complex patterns of interactions between signal and evolved effector systems are more pronounced in mammals and allow for greater plasticity in adaptive responses [1].
This chapter first discusses insulin signaling pathways and networks that are common to multiple target cell types. Recent advances in our understanding of cell processes and pathways that inhibit insulin signaling are delineated. Subsequently, unique aspects of insulin action are described, in particular key effector systems, which are properties of skeletal muscle, adipocytes, and liver, and explain the distinct effects on biologic functions in these tissues. Finally, nutrient sensing pathways and cell stress responses are discussed in terms of their interaction with insulin signaling and role in the pathogenesis of insulin resistance.
Insulin action: proximal signaling pathways
Proximal steps in insulin signaling, including the insulin receptor, insulin receptor substrate proteins (IRS), phosphatidylinositol 3 (PI3) kinase, Akt/protein kinase B (Akt/PKB), and mitogen activated protein kinase (MAPK) are globally operative in multiple cell types. These proteins also serve as points of divergence or nodes in an expanding matrix of signal transduction pathways, and are highly regulated, both positively and negatively, via cross-talk with other signaling systems and modulatory pathways (Figure 7.1).
Figure 7.1 A schematic illustration of insulin signaling pathways involved in both metabolic and mitogenic effects. Arrows represent an activation process; blocked arrows represent an inhibition process.
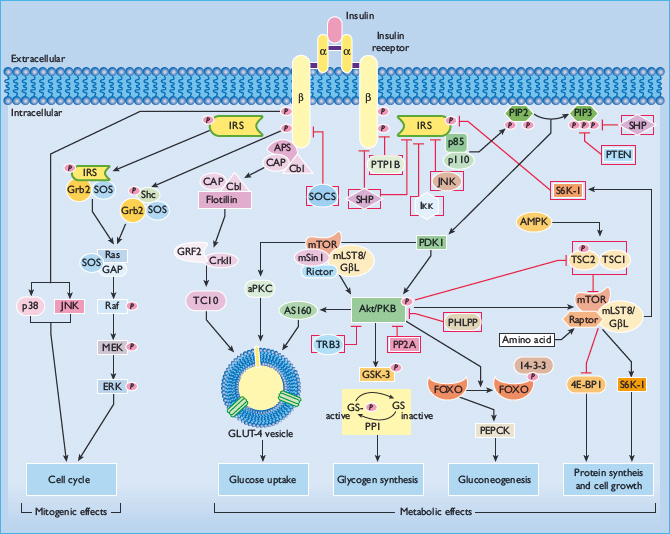
Insulin receptor molecules
Insulin action is initiated by specific binding to high-affinity receptors on the plasma membrane of its target cells. The insulin receptor is a large transmembrane glycoprotein consisting of two α- and two β-subunits which form a heterotetramer. The insulin receptor is synthesized from a single gene that consists of 22 exons and 21 introns. Following translation of its mRNA, it is processed into two separate subunits (α and β) that assemble as a disulfide-linked holoenzyme of (αβ)2 stoichiometry [2]. The 135 kDa α-subunits, derived from the amino-terminal portion of the proreceptor, reside entirely on the outside of the cell, tethered to the membrane via the 95 kDa β-subunits that span the membrane. Insulin binds to the extracellular α- subunits. This binding results in conformational changes that bring the α-subunits closer together, and enables ATP to bind to the intracellular domain of the β-subunit leading to autophosphorylation of distinct tyrosine residues on the β-subunit. Autophosphorylation augments the intrinsic activity of the β-subunit as a tyrosine kinase, directed against other tyrosines within the receptor as well as tyrosine phosphorylation of exogenous substrates. Ligand-dependent stimulation of the β-subunit tyrosine kinase activity is critical for promulgation of the insulin signal. At least six tyrosine residues in the β-subunit undergo phosphorylation and have been shown to serve different roles in insulin signaling. They lie within three functional groups. Phosphorylation of Tyr972 establishes a recognition motif and docking site that provides sufficient stability of the receptor–substrate complex for intracellular substrate phosphorylation. Tyrosine phosphorylation sites at positions Tyr1158, Tyr1162 and Tyr1163 are essential for mediating an increase in subunit tyrosine kinase activity and signal transduction. Phosphorylation sites Tyr1328 and Tyr1334 affect the sensitivity of Ras/MAPK pathway activation, and are thus involved in the receptor’s mitogenic responses [3].
The number of cell-surface insulin receptors is downregulated by chronic exposure to high insulin concentrations in vitro, and receptor loss is observed in target cells from hyperinsulinemic insulin-resistant humans. Receptor loss per se can impair maximal insulin responsiveness. This is illustrated in the extreme by genetic ablation of the insulin receptor in mice which results in lethality at 4–5 days after birth as a result of severe diabetic ketoacidosis [4]. Insulin-stimulated glucose uptake and activation of glycogen synthase in muscle are severely impaired in muscle-specific insulin receptor knockout mice [5]. These latter animals also have features of the metabolic syndrome including increases in fat mass, serum triglycerides and serum free fatty acids, but retain normal basal and contraction-stimulated glucose transport [6]. Transgenic mice expressing dominant-negative insulin receptors also develop obesity, hyperinsulinemia, glucose intolerance, and hypertriglyceridemia. These phenotypes are analogous to those seen in insulin-resistant humans. Patients with genetic mutations in the insulin receptor gene (type B insulin resistance) or circulating antibodies directed against the insulin receptor that block ligand binding (type A insulin resistance) develop severe insulin resistance, acanthosis nigricans and glucose intolerance. In addition to receptor downregulation, the intrinsic activity of the insulin receptor tyrosine kinase is impaired in insulin-resistant humans and patients with type 2 diabetes mellitus (T2DM) via a number of regulatory and pathophysiologic processes which are described in this chapter. Clearly, the number and functional activity of insulin receptors is critical for effective insulin action.
The insulin receptor is similar in structure to the insulin-like growth factor 1 receptor (IGF1R) and several other growth factor and cytokine receptors, which have in common an extracellular ligand-binding domain that activates an intracellular tyrosine kinase domain. The mammalian insulin-like signaling system includes three well-defined ligands: insulin, insulin-like growth factor 1 (IGF-1), and insulin-like growth factor 2 (IGF-2), and all three ligands can bind and activate cell-surface insulin receptors [7]. Alternative splicing involving exon 11 of the insulin receptor gene determines the insertion or deletion of 12 amino acids near the COOH-terminus of the α-subunit. Isoform A lacking the 12 amino acids has high affinity for IGF-2, predominates during fetal development, and promotes growth as a consequence of IGF-2 binding. Isoform B containing the 12 amino acids predominates postnatally and is activated mainly by insulin. Some evidence supports the contention that dysregulated expression towards the fetal pattern could occur in adult tissues and result in insulin resistance [8].
Insulin receptor substrate molecules
Following insulin binding and receptor autophosphorylation, the next committed step in signal transduction is tyrosine phosphorylation of intracellular proteins. At least 11 intracellular substrates have been identified that are rapidly phosphorylated on tyrosine residues by ligand-bound insulin receptors, including six insulin receptor substrate (IRS) proteins, Grb2-associated binder 1 (Gab1), Cas-Br-M (murine) ecotropic retroviral transforming sequence homolog (Cbl), and the various isoforms of Src-homology-2-containing protein (Shc) [9].
The IRSs are immediate substrates for the insulin receptor tyrosine kinase, and are of predominant importance for insulin action. IRS proteins have an N-terminal pleckstrin-homology (PH) domain, a phosphotyrosine-binding (PTB) domain and a COOH-terminal region of variable length that contains multiple tyrosine and serine phosphorylation sites [10]. The PH domain helps position IRS for coupling with the insulin receptor, possibly by binding to charged headgroups of certain phosphatidylinositides in adjacent membrane structures. PTB domains recognize the phosphotyrosine in the amino acid sequence asparagine-proline-any amino acid-phosphotyrosine (NPXpY), which encompasses Tyr972 in the juxtamembranous domain of the insulin receptor β-subunit, and facilitates the formation of the IRS–insulin receptor complex. The center and C-terminus of IRS proteins contain up to 20 potential tyrosine phosphorylation sites that, after phosphorylation by the insulin receptor, bind to other intracellular molecules that contain Src-homology-2 domains (SH2 domains). The SH2-containing proteins that bind to phosphorylated IRS proteins could be adaptor molecules, such as the regulatory subunit of PI3 kinase and the adaptor molecule Grb2, or enzymes with kinase or phosphatase activities such as SH2-domain-containing tyrosine phosphatase 2 (SHP2) and the cytoplasmic tyrosine kinase Fyn. Non-covalent attachment of multiple SH2-containing signaling molecules allows IRS to function as a docking protein that physically apposes proteins involved in downstream transduction cascades. Thus, IRS propagates insulin signal transduction via the docking, apposition, interaction, and activation of downstream signal molecules, rather than as a consequence of any intrinsic enzymatic activity per se. Thus, IRSs provide a major point of divergence of insulin signal transduction pathways, for example, leading to activation of mitogenic (Ras/MAP kinase) and metabolic (PI3 kinase) pathways [11].
Among the family of IRS proteins, IRS1 and IRS2 exhibit a wide range of tissue expression, including muscle, fat, liver, and pancreatic islets. IRS3 is expressed in adipose tissue, fibroblasts, and liver, while IRS4 is detected in brain, thymus, and embryonic kidney. IRS5 is ubiquitously expressed but most abundant in kidney and liver. IRS6 expression is highest in skeletal muscle. Deletion of IRS1 produces small insulin-resistant mice with nearly normal glucose homeostasis because of β-cell expansion and compensatory hyperinsulinemia. Mice lacking IRS2 display nearly normal growth but develop life-lhreatening diabetes at 8–15 weeks of age because of reduced β-cell mass and insufficient compensatory insulin secretion. IRS3 knockout mice do not have an obvious phenotype, and IRS4 null mice also appear normal with the exception of reduced fertility. Therefore, IRS1 and IRS2 are considered to be the IRS isoforms critically important in glucose homeostasis. IRS1 functions as the principal IRS in skeletal muscle, and IRS2 predominates in liver and β-cell, where insulin action is required for normal β-cell growth and development.
IRS proteins are a key locus for regulation of insulin action. One mechanism occurs at the level of IRS protein expression; for example, IRS1 and IRS2 proteins are decreased by hyperinsulinemia [12]. Cell loss of IRS could occur through accelerated protein degradation by induction of ubiquitin-mediated degradation of IRS1 and IRS2 by suppressor of cytokine signaling (SOCS) proteins [13], or by inhibition of IRS gene transcription. Regardless of the mechanism, decreased levels of IRS proteins in hyperinsulinemic states, coupled with downregulation of the insulin receptor itself, certainly contribute to the insulin resistance in diabetes [14]. The function of IRS proteins can also be negatively regulated by serine–threonine kinases and protein tyrosine phosphatases such as such as PTP1B and SHP2, as described below [15]. In addition, IRS1 can be post-translationally modified by either O-linked N-acetylglucosamine adducts (O-GlcNAc) on serine–threonine residues under hyperglycemic conditions [16], or S-nitrosylation as a consequence of nitric oxide generation [17]. These modifications induce the proteasomal downregulation of IRS1 and insulin resistance.
PI3 kinase
Among several proteins that to bind to IRS1/2, PI3 kinase is critical for signal transduction mediating the metabolic effects of insulin, including stimulation of glucose uptake into skeletal muscle and adipose tissues. PI3 kinase activity represents a family of related enzymes that are capable of phosphorylating the hydroxyl groups in the inositol ring of membrane-associated phosphatidylinositol (PtdIns) [18]. The generation of PI(3,4,5)P3 propagates the insulin signal, and therefore the class I subset of PI3K is responsible for the downstream action of receptor tyrosine kinases (including the insulin receptor) and Ras/MAPK [19]. Class I PI3 kinases are heterodimeric molecules composed of one regulatory and one catalytic subunit. There are currently five known regulatory subunits, designated p85α, p55α, p50α, p85β or p55γ (known collectively as the p85 subunit), and one of these regulatory subunits is conjoined with one of four known p110 catalytic subunits, p110α, p110β, p110δ or p110γ [20]. Under normal conditions, p85 regulatory subunits are present in excess compared with the amount of the p85–p110 complex, and can serve as negative regulators of insulin action. One explanation pertains to the stoichiometry of free p85 and p85–p110 complexes because free p85 may be able to compete with the p85–p110 complex for recruitment to IRS phosphotyrosine docking sites (i.e. the “free p85” model) [21]. Accordingly, increased expression of p85 can worsen insulin sensitivity as demonstrated in patients with gestational diabetes or obesity who have increased levels of p85 in skeletal muscle. The p85 subunit can also exert negative modulatory effects via cross-talk with stress-kinase pathways. Recent studies have shown that p85 is required for the insulin-stimulated activation of c-Jun NH2-terminal kinase (JNK) that occurs in states of insulin resistance, including high-fat diet-induced obesity and JNK overexpression [22]. Because the JNK pathway suppresses insulin action, the involvement of p85 in JNK activation provides a mechanism for cross-talk between the PI3 kinase signaling pathway and JNK-mediated stress or inflammatory responses. Also, p85 is able to suppress insulin action via positive regulation of the phosphatase and tensin homolog (PTEN), a phosphoinositide phosphatase that degrades PI(3,4,5)P3 and inhibits downstream insulin signaling [23].
3-Phosphoinositide-dependent protein kinase 1
Insulin-mediated activation of PI3 kinase results in phosphorylation of the inositol ring at the 3’ position of phosphatidylinositol in membrane glycolipids, generating PI(3,4,5)P3. This leads to recruitment of certain signaling proteins with PH domains to the plasma membrane. Binding to membrane-associated phosphoinositides both activates these proteins and positions them for downstream signal transduction. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) can interact with PI(3,4,5)P3, and is responsible for downstream activation of Akt/PKB and aPKCs. PDK1 phosphorylates the activation loops of Akt/PKB on Thr308, and PKCζ on Thr410, enhancing the activity of these kinases.
Akt/protein kinase B
Phosphorylation and activation of Akt/PKB mediates various insulin- and growth factor-induced cellular responses, such as the stimulation of GLUT-4 translocation to the plasma membrane, the inhibition of glycogen synthase kinase 3 (GSK-3), induction of triglyceride synthesis via increasing the expression of sterol regulatory element-binding proteins 1c (SREBP-1c), and the promotion of cell survival by inhibiting apoptosis. Akt/PKB is a serine–threonine kinase with multiple substrates including kinases, signaling proteins, and transcription factors, such as cyclin-dependent kinase inhibitor p21kip, GSK3β, Bcl-2 antagonist of cell death, AS160, endothelial NO synthase, forkhead box class O1 (Foxo 1) and others. There are three isoforms of Akt/PKB in mammals, each encoded by a different gene (Akt1, Akt2 and Akt3). Akt1 and Ak2 are widely distributed; however, Akt2 is predominant in insulin-sensitive tissues such as liver and fat. Akt3 is largely expressed in the nervous system and testis.
Several lines of evidence implicate a role for Akt/PKB activation in the stimulation of glucose transport and other insulin-induced biologic processes. Expression of constitutively active membrane-bound forms of Akt/PKB results in persistent translocation of GLUT-4 to the plasma membrane in muscle and fat cells [24]. Deletion of Akt1 results in growth retardation and reduced lifespan without metabolic abnormalities [25]. In contrast, Akt2-deficient mice display insulin resistance and develop diabetes as a result of the inability of insulin to stimulate glucose utilization and decrease hepatic glucose output [26]. Mechanistic studies have suggested that Akt2 is the key isoform transducing effects on GLUT-4 glucose uptake; however, Akt/PKB activity alone cannot fully explain the ability of insulin to promote GLUT-4 translocation and stimulate glucose transport [27].
Akt/PKB activation is involved in multiple other insulin responses. One of the first substrates indentified for Akt/PKB was GSK3β. Phosphorylation of GSK3β decreases its activity towards glycogen synthase, which leads to increased glycogen synthesis [28]. Akt/PKB seems to stimulate glucose transport concomitant with the phosphorylation of Akt substrate of 160 kDa (AS160), a Rab-GTPase-activating protein (GAP) [29]. Under basal conditions, a functional GAP domain within AS160 is necessary to maintain the intracellular localization of GLUT-4 [30]. Following insulin stimulation, phosphorylation of AS160 shuts off its GAP activity, shifting the equilibrium of its target Rab(s) to an active GTP-bound form, and this enables GLUT-4 translocation by releasing GLUT-4 from intracellular retention mechanisms [30]. As expected, this effect is blocked by pre-exposure to the PI3 kinase inhibitor wortmannin, indicative of signaling through PI3 kinase. This response involving AS160 is impaired in skeletal muscle collected from insulin-resistant patients.
Atypical protein kinase C
Protein kinase C isoforms are categorized as conventional (α,β,γ) (cPKC), novel (δ,θ,ε,η,μ) (nPKC), and atypical (ζ,λ) (aPKC) depending on their ability to be activated by calcium and diacylglycerol (DAG). cPKCs and nPKCs serve primarily as negative feedback inhibitors of the insulin receptor and IRS, and are discussed below. The aPKCs participate in signal transduction, and like Akt/PKB, are activated by the PI3 kinase pathway and help mediate insulin’s metabolic effects. The two aPKC isoenzymes ζ and λ vary in their tissue-specific expression and among various mammals; for example, PKCλ is the main aPKC in the skeletal muscle and adipose tissue of the mouse, whereas PKCζ is more prominent in rats, monkeys, and humans. Activation of PKCζ and PKCλ occurs proximal to Akt/PKB as a result of direct interaction with 3’ phosphoinositides and/or through phosphorylation and activation by PDK1. aPKCs have been shown to have a role in insulin-stimulated glucose uptake and GLUT-4 translocation in adipocytes and muscle [31]. Stimulation of glucose transport and translocation of GLUT-4 vesicles by aPKC may be brought about by effects on the actin cytoskeleton, because PKCλ/ζ can impinge on Rac and actin dynamics [32]. Overexpression of constitutively active forms of PKCζ and PKCλ increases, and expression of dominant negative forms of these aPKCs reduces, both glucose transport activity and GLUT-4 translocation in response to insulin (31). Decreased activation of aPKCs has been reported in the muscle of humans with T2DM and rodents with insulin resistance [33]. Compellingly, a mouse model with a muscle-selective PKCλ gene deletion displays whole-body insulin resistance, impaired insulin-stimulated glucose uptake into muscle and reduced GLUT-4 translocation. The mechanism by which aPKC participates in metabolic signaling remains to be fully elucidated.
CAP/Cbl/TC10 pathway
In parallel to the PI3 kinase pathway, substantial evidence has confirmed the participation of the CAP/Cbl/TC10 pathway in glucose transport stimulation [34]. This pathway diverges at the level of the insulin receptor kinase, which mediates tyrosine phosphorylation of the Cbl proto-oncogene through a process that does not involve IRSs. This phosphorylation step requires recruitment of the adapter protein APS, which contains SH2 and PH domains, to the insulin receptor β-subunit and the subsequent binding of Cbl to APS [35]. APS interacts via its SH2 domain with phosphotyrosines in the activation loop of the insulin receptor. Upon binding to the receptor, APS is phosphorylated on a C-terminal tyrosine, which permits the recruitment of Cbl to APS via the SH2 domain of Cbl, and subsequent tyrosine phosphorylation of Cbl. The Cbl associated protein (CAP) is also recruited with Cbl to the insulin receptor–APS complex via tandem SH3 domains in the COOH-terminus of CAP that bind to a proline-rich domain in Cbl. CAP also contains a sorbin homology (SoHo) domain in the NH2-terminal region. After disengagement of CAP/Cbl from the insulin receptor, the CAP SoHo domain binds to flotillin in caveolin-containing lipid rafts in the plasma membrane. Lipid rafts are plasma membrane domains, enriched in cholesterol, glycolipids, and sphingolipids, that coordinate signaling events by accumulating specific protein constituents.
Once the CAP/Cbl complex is bound to flotillin, tyrosine phosphorylated Cbl presents a recognition site for recruitment of the CrkII–C3G complex to the lipid raft. CrkII binds to specific phosphorylation sites on Cbl via its SH2 domain and is constitutively associated with the nucleotide exchange factor C3G via its SH3 domain. C3G is a guanyl nucleotide exchange factor for TC10 and other small molecular weight GTP-binding proteins. TC10 is a member of the Rho family of GTPases, and can target to lipid raft domains as a result of its capacity to undergo post-translational modification by farnesylation and palmitoylation. The Rab proteins cycle between GTP- and GDP-bound states to affect vesicle budding from donor membranes and fusion with acceptor membranes. TC10 is known to regulate the actin cytoskeleton [36], and downstream effectors of TC10 have been identified which have been proposed to have a role in the translocation of GLUT-4 [37].
Ras-p38 MAPK pathway
Another component of signal divergence emanating from IRS docking proteins is the engagement of the Ras/MAPK signaling pathway, which has a critical role in cell growth and mitogenesis. Following insulin-mediated tyrosine phosphorylation of IRS, one of the SH2 domain-containing proteins that docks with IRS is Grb-2, a small cytosolic adapter protein. Grb-2 also contains an SH3 binding domain that binds proteins containing proline-rich sequences, and one of these proteins is SOS (mammalian homolog of the Drosophila son-of-sevenless protein), a GDP/GTP exchange factor. This interaction positions SOS for activation of the Ras/MAPK pathway. SOS facilitates GTP activation of membrane-bound Ras, the 21 kDa small molecular weight GTPase, and the GTP-bound form of Ras complexes with and activates Raf-1 kinase. Raf-1 kinase then initiates a cascade leading to sequential phosphorylation and activation of the dual-specificity kinase MEK (MAPK/ERK kinase), which in turn phosphorylates extracellular regulated kinases (ERK1 and ERK2) on threonine and tyrosine residues in their activation loops. Activated ERKs phosphorylate multiple targets that mediate the mitogenic actions of the Ras/MAPK pathway and the growth promoting effects of insulin [38]. Insulin receptors can also mediate activation of the Ras/MAPK pathway through another substrate docking molecule, SHC. Independent of IRS, SHC can also activate the Ras/Raf-1/MEK phosphorylation cascade by forming a complex with Grb-2/SOS in response to insulin. Whether activated through IRS or SHC, ERK1/2 translocates into the nucleus and phosphorylates transcription factors, such as ELK-1, thus modulating DNA binding properties and regulation of gene transcription. ERK also phosphorylates p90 ribosomal protein S6 kinase (p90 S6 kinase), which can phosphorylate and regulate the activity of transcription factors such as c-fos. In addition, the MAPK cascade is one of the pathways with the potential to stimulate glycogen synthase because p90 S6 kinase is able to activate the glycogen-associated protein phosphatase-1, which in turn dephosphorylates and activates glycogen synthase [39]. In this way, the MAPK pathway has the potential to interact with metabolic signaling pathways.However, the MAPK pathway is not necessary for stimulation of glucose transport, and is not viewed as being critically related to the metabolic effects of insulin [40].
Inhibition of insulin signal transduction
The promulgation of insulin signaling pathways does not proceed unabated; rather, there is an extensive array of mechanisms that dampen or inhibit signal transduction. These inhibitory mechanisms can represent normal dynamic functioning of insulin action as organisms adapt to changing physiologic conditions, or, when unbalanced, can lead to pathophysiologic consequences and the development of insulin resistant states. The elucidation of inhibitory processes has provided insight into the extensive network of regulated insulin action pathways, and has also identified potential therapeutic targets because blocking these inhibitory mechanisms could enhance insulin sensitivity.
Inhibition of the insulin receptor
As the first critical component in the insulin signaling network, there are also multiple mechanisms that can desensitize insulin action at the level of the insulin receptor. For example, it has long been known that persistent insulin stimulation can lead to loss of cell surface insulin receptors and this downregulation event impairs insulin sensitivity. In addition, multiple cellular proteins and processes can negatively regulate the intrinsic tyrosine kinase activity of the insulin receptor. Two mechanisms (serine–threonine phosphorylation and protein tyrosine phosphatases) act to impair both insulin receptors and IRS docking molecules.
Downregulation of insulin receptor number
Chronic exposure to high insulin concentrations leads to loss of insulin receptors, resulting in a rightward shift in insulin dose–response curves (i.e. impaired insulin sensitivity). This is caused by increased ligand-mediated internalization of insulin receptors followed by lysosomal degradation, as well as diminished gene expression. Insulin stimulation results in phosphorylation of Foxo1, which disrupts its interaction with the insulin receptor promoter, decreasing insulin receptor gene transcription, thus contributing to receptor loss and insulin resistance [41].
Plasma differentiation factor 1
Plasma differentiation factor 1 (PC-1), also referred to as ectonucleotide pyrophosphatase phosphodiesterase 1, is a membrane glycoprotein with pyrophosphatase activity that appears to act as an intrinsic inhibitor of the insulin receptor tyrosine kinase [42]. PC-1 binds to amino acids 485–599 of the insulin receptor connecting domain, a region required for the conformational change in receptor β-subunits that permit autophosphorylation upon insulin binding. This interaction with PC-1 interferes with the close opposition of the two β-subunits required for transphosphorylation. Muscle expression of PC-1 is elevated in patients with diabetes and in obesity, and it correlates with diminished insulin receptor tyrosine phosphorylation and muscle glucose uptake. Analysis of PC-1 gene polymorphisms in a variety of individuals and family cohorts suggests that alterations in this gene are associated with the risk for development of childhood and adult obesity as well as T2DM [43].
Grb proteins
The growth factor receptor-bound proteins (Grb proteins) constitute a family of structurally related multi-domain adapters with diverse cellular functions but lacking intrinsic enzymatic activity. Grb10 and Grb14 can bind to phosphotyrosine residues on the insulin receptor and alter receptor tyrosine kinase activity [44]. Overexpression of Grb10 and Grb14 in cells inhibits insulin-stimulated phosphorylation of IRS1, IRS2 and Shc [45]. However, the physiologic role of Grb proteins is not fully clear, and their actions may be tissue specific with capabilities as an inhibitory factor or a positive mediator in the insulin signaling pathway.
Inhibition of insulin receptor substrate proteins
Protein phosphotyrosine phosphatases
Endogenous protein phosphotyrosine phosphatases (PTPases) are able to dephosphorylate tyrosine residues on the insulin receptor β-subunit and insulin receptor substrate docking molecules, resulting in a dampening of insulin signal transduction. Two PTPases in particular, PTP-1B and leukocyte common antigen-related phosphatase (LAR), contribute to insulin receptor dephosphorylation in insulin target cells [46]. Membrane-associated PTPase activity is increased in skeletal muscle from patients with T2DM [47], principally because of increments in cytosolic PTPase-1B and membrane-associated LAR [48]. The ability of PTPases to modulate insulin signaling has been demonstrated in mice with genetic ablation of PTPase-1B, which exhibit enhanced insulin sensitivity, increased insulin-mediated tyrosine phosphorylation of the receptor and of IRS1, and a failure to develop insulin resistance when fed a high fat diet [46]. Muscle-specific overexpression of PTP-1B in mice induces tissue insulin resistance with decreased capacity for insulin receptor autophosphorylation [49]. Thus, available data consistently demonstrate that PTPase-1B is able to modulate insulin signaling negatively through dephosphorylation of tyrosine residues in the insulin receptor and IRSs. The role of the LAR in insulin receptor function remains less well defined.
serine–threonine phosphorylation
serine–threonine phosphorylation of insulin receptors and IRS docking proteins is a major mechanism for negative modulation of insulin signal transduction. Serine phosphorylation diminishes insulin receptor tyrosine kinase activity and decreases receptor–IRS coupling by inhibiting insulin-mediated tyrosine phosphorylation of IRS-1, binding and activation of PI3 kinase, and stimulation of glucose transport. There are consensus sequences in IRS-1 that make it susceptible to a wide variety of serine–threonine kinases including PKC, PKA, Akt/PKB, MAPK, S6 kinase, GSK3, casein kinase II, Cdc2 kinase, JNK and IκB kinase (IKKβ). Several of these kinases have been shown to function as physiologic modulators causing desensitization of insulin signaling pathways under conditions of nutrient excess, inflammation and cell stress responses. For example, JNK and IKKβ are activated by inflammatory stimuli (e.g. TNFα) contributing to insulin resistance, and PKC is activated by DAG which accumulates with increased availability of free fatty acids.
Protein kinase C
PKCs are serine–threonine kinases with multiple substrates, including IRS docking proteins and the insulin receptor [50]. Serine–threonine phosphorylation of IRS impairs its ability to associate with the insulin receptor and with PI3 kinase, resulting in desensitization of the PI3 kinase pathway. Hyperinsulinemia, hyperglycemia and elevated circulating free fatty acids (e.g. nutrient excess) leads to increased intracellular DAG, which in turn activates conventional and novel PKC isoforms principally via recruitment to the plasma membrane. These conditions are associated with increased serine–threonine phosphorylation and diminished function of insulin receptors and IRS proteins. In addition, insulin activates atypical PKCs, such as PKCζ, via the PI3 kinase pathway, which is also capable of phosphorylating and desensitizing IRS [51].
Tumor necrosis factor α
Tumor necrosis factor α (TNF-α) is a cytokine produced by immune cells and also by adipocytes and muscle tissue. While having little impact on systemic circulating concentrations, TNF-α expression is increased in adipose and muscle tissues as a function of insulin resistance [52]. In adipose tissue, TNF-α and other proinflammatory cytokines are produced by adipocytes and also by macrophages that infiltrate adipose tissue under conditions of obesity and insulin resistance. This raises the possibility that the cytokine could be inducing cellular insulin resistance via autocrine and/or paracrine effects. TNF-α induces serine phosphorylation of IRS1, thereby decreasing its ability to be phosphorylated by the insulin receptor tyrosine kinase and impairing downstream insulin signal transduction [53]. Several pathways activated by TNF-α are implicated in increased serine phosphorylation of IRS-1, including the stress-induced kinases, JNK and IKKβ.
JNK
Three JNK-encoding genes have been described in mammals; JNK1 and JNK2 are expressed ubiquitously, whereas JNK3 expression is restricted to neuronal tissues. The JNK isoforms belong to the extended family of MAPKs, and control many cellular functions through regulation of activator protein 1 (AP-1). In addition to TNF-α signaling, insulin also activates JNK1 and JNK2 [54], which then display increased serine kinase activity against multiple intracellular substrates including IRS1, IRS2 and Shc. The ability of insulin to activate JNK represents a negative feedback mechanism by which insulin inhibits its own signaling.
JNK activity is also augmented during cellular stress responses, such as endoplasmic reticulum stress, and is increased in insulin-resistant states. Illustrative data include the observation that high-fat diets increase Ser307 phosphorylation of IRS1 in wild-type mice but not in JNK1–/– mice, while the JNK1–/– mice are characterized by decreased adiposity, increased insulin receptor signaling and improved insulin sensitivity [55]. Thus, JNK has been implicated in the pathogenesis of insulin resistance in the metabolic syndrome and T2DM.
NF-κB
Activation of NF-κB-mediated pathways has been shown to inhibit insulin signaling through enhanced serine phosphorylation of IRS1 [56]. NF-κB is a transcription factor that functions as a proinflammatory “master switch” during inflammation, upregulating the transcription of a wide range of inflammatory mediators. NF-κB is normally retained in the cytoplasm by binding to members of the inhibitor of κB (IκB) protein family. During inflammatory or metabolic stress, NF-κB is activated by the IκB kinase (IKK) complex, which consists of two catalytic subunits, IKKα and IKKβ and a regulatory subunit, IKKγ. The IKKβ subunit phosphorylates IκB, resulting in its ubiquitination and subsequent proteasomal degradation and in the release of NF-κB. The free NF-κB is then able to translocate into the nucleus and activate the transcription of at least 125 genes, most of which are proinflammatory [57]. There exists a substantial body of data implicating NF-κB in the pathogenesis of insulin resistance. Activation of NF-κB accompanies insulin resistance following high fat feeding and obesity, and in the metabolic syndrome. Proinflammatory cytokines such as TNF-α and interleukin 6 (IL-6) can also activate NF-κB. Circulating mononuclear cells in the obese display increased transcription of proinflammatory genes regulated by NF-κB with a decrease in IκB. Weight loss, caloric restriction and exercise training can lead to a reduction in transcription of the proinflammatory genes regulated by NF-κB [58]. Drugs with anti-inflammatory properties, such as thiazolidinediones, statins and salicylates, can inhibit proinflammatory cytokine secretion by interferring with the NF-κB pathway.
IκB kinase-β
IKKβ is a serine kinase that has the ability to desensitize insulin signaling through serine phosphorylation of IRS or the insulin receptor. The desensitizing effect of TNF-α, as a consequence of IRS serine phosphorylation, may in part be mediated through IKKβ. Consistent with this hypothesis, heterozygous ablation of the IKKβ gene in mice fed a high-fat diet, or in obese leptin-deficient ob/ob mice, prevents insulin resistance [59,60]. Mice that selectively express constitutively active IKKβ in hepatocytes (LIKK mice) exhibit a T2DM phenotype, characterized by hyperglycemia, profound hepatic insulin resistance, and moderate systemic insulin resistance including effects in muscle [56]. The hepatic production of proinflammatory cytokines, including IL-6, IL-1β and TNF-α is increased in LIKK mice to a similar extent as induced by high-fat feeding in wild-type mice. However, no difference in obesity-induced insulin resistance is detectable in muscle-specific IKKβ knockout mice compared with wild type [61], which suggests that IKKβ may be more directly involved in the development of hepatic insulin resistance rather than in skeletal muscle.
An important biologic effect of salicylates is the inhibition of the IKKβ[62]. Predictably, salicylates would then enhance insulin sensitivity by causing a subsequent decrease in IKKβ-mediated serine phosphorylation of IRS. In fact, treatment with high doses of salicylates improves glucose tolerance and enhances insulin sensitivity in humans and rodents [59].
Accelerated catabolism of PI(3,4,5)P3 by phosphoinositide phosphatases
Increased production of PI(3,4,5)P3 as a result of activated PI3 kinase is key to metabolic insulin signaling, and the downstream activation of PDK1. PTEN can dephosphorylate PI(3,4,5)P3 on the 3 position, and SH2-containing inositol phosphatases (SHIPs) can dephosphorylate PI(3,4,5)P3 on the 5 position of the inositol ring; in either instance, the ability of PI(3,4,5)P3 to stimulate PDK1 is lost. Therefore, PTEN and SHIP can exert negative regulatory influences on insulin signaling, particularly with respect to its metabolic actions [63]. While multiple studies support the view that PTEN can participate as a negative regulator of insulin action in pathophysiologic states, its role in human insulin resistance is yet to be defined.
Inhibition of insulin signaling by protein–protein interactions
Suppressor of cytokine signaling 3
The SOCS family of proteins (CIS and SOCS 1–7) was originally described as a negative feedback loop for cytokine receptors involving Janus kinase (JaK). Following ligand activation of cytokine and growth factor receptors, JaK phosphorylates and activates signal transducer and activator of transcription (STAT), and these phosphorylated STAT family members then form homodimers or heterodimers that translocate to the cell nucleus where they act as transcription activators. In the nucleus, STAT augments SOCS gene expression. SOCS proteins then feedback to inhibit tyrosine-phosphorylated cytokine receptors, via either competitive binding through their SH2 domain preventing phosphorylation of cytokine receptor substrates, or by binding and inhibiting the action of JaK tyrosine kinases. Evidence suggests that a similar mechanism may be operative for the insulin receptor [46]. Studies have indicated that SOCS-1, SOCS-3 and SOCS-6 can bind to the COOH-terminus of the insulin receptor β-subunit, and block interaction between the insulin receptor and IRS [64]. Interestingly, several factors that induce cellular insulin resistance also induce SOCS-3 expression, including TNF-α, growth hormone, and leptin.
Tribbles
The Tribbles (TRB) gene family in mammals is comprised of three proteins that have a truncated kinase domain lacking an ATP binding site. Accordingly, TRBs are “pseudokinases” that lack detectable kinase activity, but can bind to kinase substrates in phosphorylation cascades and inhibit their phosphorylation. TRB proteins, for example, can bind to Akt/PKB and inhibit its phosphorylation and activation in response to insulin [65], while downregulation of TRB3 improves insulin sensitivity [66].
Tissue-specific insulin action: the role of insulin effector systems
Insulin regulates whole-body fuel homeostasis via specific effects in multiple target tissues. The nature of these biologic actions varies dramatically from tissue to tissue, and these variations, for the most part, are not brought about by differences in insulin signal transmission (described above). Rather, tissue-specific insulin effects are principally explained by effector systems that are uniquely expressed in a variety of differentiated target cells. The biochemical basis of these effects is described in skeletal muscle, adipose tissue, and liver, three organs primarily responsible for fuel storage and oxidation as well as counter-regulatory metabolism.
Skeletal muscle
Insulin stimulation of glucose transport
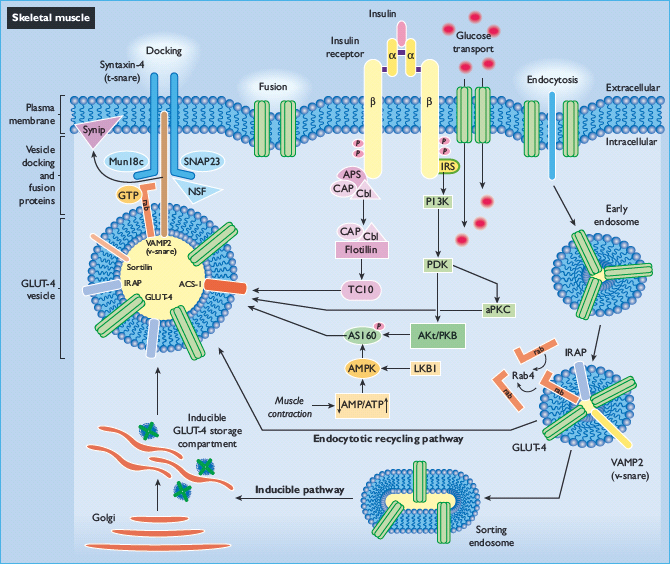
Skeletal muscle accounts for the bulk of insulin-stimulated glucose uptake in vivo, and the hallmark of insulin action in this tissue is the ability to stimulate the glucose transport effector system (Figure 7.2). Glucose transport across the plasma membrane is facilitated by glucose transport proteins (GLUT); to date, 13 members of the GLUT/SLC2 family have been identified. All GLUT proteins are intimately embedded in membranes, and the most highly conserved regions are the putative membrane-spanning domains that serve a common function, the creation of a pore for facilitative diffusion of monosaccharides. Each glucose transporter isoform has a specific role in glucose metabolism determined by its pattern of tissue expression, substrate specificity and affinity, transport kinetics, and regulated expression in different physiologic conditions.
The major transporter isoforms that mediate glucose transport in cells with an insulin-responsive glucose transport system are GLUT-1 and GLUT-4. In unstimulated cells, GLUT-1 predominates at the cell surface and facilitates glucose diffusion across the plasma membrane into the cytosol where glucose is rapidly phosphorylated by hexokinase and metabolized. GLUT-4 contributes minimally to glucose transport in unstimulated target cells, because >90% of the cell content of GLUT-4 resides in intracellular membranes in the basal state. The mechanism by which insulin augments glucose transport activity is by recruiting intracellular GLUT-4 to the plasma membrane, a rate-limiting step for insulin-stimulated glucose uptake and metabolism in peripheral target tissues. Upon dissipation of the insulin signal, deactivation of glucose transport activity is the result of a net reverse translocation of GLUT-4 transporters back into the cell interior. Thus, GLUT-4 is the major transporter mediating insulin-stimulated glucose transport activity in tissues such as skeletal and cardiac muscle and adipose tissue.
In unstimulated muscle or adipose cells, a component of GLUT-4 resides in an inducible tubulo-vesicular storage compartment that includes the trans-Golgi network and endosomal vesicles located near the endofacial surface of the plasma membrane. However, another component of cellular GLUT-4 exists in an active endocytosis–endosomal recycling pathway that cycles GLUT-4 between endosomes and the plasma membrane. The recycling pathway results in the localization of approximately 4–10% of GLUT-4 in the basal plasma membrane, and this steady-state distribution is the balance of rapid endocytosis and slow recycling. Insulin shifts the distribution of GLUT-4 from intracellular pools towards the plasma membrane, both by elevating the exocytotic rate of GLUT-4 in the recycling pathway and by recruiting GLUT-4 from the inducible storage compartment to the cell surface. Deactivation of transport is accomplished via a slowing of the exocytotic rate and an acceleration of the endocytotic rate, as GLUT-4 is retrieved from the plasma membrane through clathrin-dependent and -independent mechanisms [67,68].
GLUT-4 vesicle trafficking involves the actin and microtubule cytoskeletons. Regarding actin, insulin stimulates cytoskeletal rearrangement with the appearance of cortical β-actin fiber projections that subtend the plasma membrane, and this actin remodeling is under the control of small G-proteins in the Rho, Rab and Rac families. Microtubules surround the inducible intracellular depot of GLUT-4 and microtubule proteins such as dynein and kinesin have been co-purified with GLUT-4. Inhibition of actin remodeling or disruption of microtubules using depolymerizing agents inhibits GLUT-4 translocation and glucose transport stimulation. Data suggest that exocytotic movement of GLUT-4 begins with its transfer to actin scaffolds that connect the microtubule cytoskeleton with the plasma membrane, which positions GLUT-4 vesicles for docking and membrane fusion.
The complete pathway linking insulin signal transduction to stimulation of the glucose transport system has not been fully elucidated. Activation of transport is critically dependent on insulin-mediated autophosphorylation of insulin receptors, tyrosine phosphorylation of IRS1 and activation of PI3 kinase. The production of PI(3,4,5)P3 by PI3 kinase activates PDK1. At this point, PDK1 activates two separate kinase pathways that contribute to GLUT-4 translocation and stimulation of glucose transport activity, Akt2 and atypical PKCs (PKCζ and PKCλ). Akt2 then phosphorylates and inhibits AS160 (TBC1D4) and TBC1D1 [69], which are Rab-GTPase-activating proteins. The modulation of AS160 activates Rab small GTPases that in turn regulate aspects of GLUT-4 vesicle docking and cytoskeletal organization [70]. At the same time, the ligand-bound insulin receptor activates the CAP/Cbl/TC10 pathway upstream of IRS1 phosphorylation, and activates TC10, a Rho family member that regulates the actin cytoskeleton. Importantly, the factors that link TC10, AS160, atypical PKCs, and other relevant factors with the glucose transport effector system, and which impel the complex changes in GLUT-4 vesicle trafficking associated with transporter translocation, are unknown.
Figure 7.2 An overview of insulin signaling pathways in skeletal muscle. Arrows represent an activation process.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


