Inhibitors of Fibrinolysis
BjÖRn Wiman
Anna ÅGren
The fibrinolytic system has many different functions in vivo. Within the blood vessels, its main function is to remove fibrin deposits and keep the blood vessels open after clots have been formed. But the fibrinolytic system also seems to play roles in other processes such as reproduction, tissue repair and remodeling, invasive growth, and metastasis.1 When activated, the fibrinolytic system results in the formation of active plasmin, a very powerful proteolytic enzyme, with an action very similar to that of trypsin. Plasmin eventually has the capacity to degrade almost all of the plasma proteins in vivo. It is therefore of utmost importance that both its action and its formation are highly regulated.2 This is achieved in two distinct ways. Firstly, activation of plasminogen to plasmin physiologically occurs preferentially at the surface of fibrin. Secondly, free plasmin becomes inactivated rapidly. In that way, the action of the fibrinolytic system in the blood vessels is localized to eventual deposits of fibrin. Fibrinolytic inhibitors play important regulatory roles in different stages of this process. The inhibitors are mainly antiplasmin and PAI-1 (plasminogen activator inhibitor 1), two inhibitors belonging to the serine proteinase inhibitors or the serpin superfamily of proteins. In addition, other substances influence the process both in a positive and a negative way. They may also be of great physiologic significance. A schematic picture of the fibrinolytic process and its regulation is demonstrated in FIGURE 21.1. In this chapter, we mainly discuss the inhibitors involved in this regulation, such as those inhibiting plasmin, which are antiplasmin and α2-macroglobulin, those inhibiting plasminogen activators, which are PAI-1 and PAI-2 (plasminogen activator inhibitor 2), and finally, other compounds influencing the process, such as fibrin and TAFI (thrombin-activatable fibrinolysis inhibitor).
From the pathophysiologic point of view, an increased function of the fibrinolytic system leads to an increased bleeding tendency. This is, for example, seen in patients with antiplasmin deficiency and may result in bleedings as severe as in patients with hemophilia. On the other hand, a decreased fibrinolytic function, as is found in patients with increased plasma levels of PAI-1, gives a thrombotic tendency both in the arteries and in the veins.
ANTIPLASMIN
Discovery and Basic Information
Antiplasmin is a serpin with a molecular weight of about 70,000. It was discovered in 1976 by three different groups.3,4,5 It is produced in the liver,6 and therefore its plasma concentration is decreased in severe liver diseases where protein biosynthesis is diminished.7 The plasma concentration of antiplasmin is about 70 mg/L (1 µmol/L), which is approximately half the concentration of plasminogen in plasma on a molar basis. Therefore, under normal circumstances, when only little plasminogen is activated, the antiplasmin concentration is in far excess of the formed plasmin. However, during thrombolytic treatment, especially with non-fibrin-specific plasminogen activators, such as streptokinase or urokinase, large amounts of the plasminogen will become activated rapidly with the risk of all the antiplasmin becoming depleted. Under those circumstances, α2-macroglobulin might become an important inhibitor of plasmin. The half-life in the circulation of native antiplasmin is approximately 3 days.8
Antiplasmin has several different functional properties. Its main physiologic activity is inhibition of plasmin by forming an irreversible enzymatically inactive complex with plasmin.9,10 It inhibits plasmin extremely rapidly.9 In addition, it inhibits the binding of plasminogen to fibrin, which is a very important regulatory phenomenon in the fibrinolytic process.2,11 It may also become cross-linked to the α-chains of fibrin during clotting by the action of coagulation factor XIIIa.12,13 Antiplasmin bound to fibrin in this way is believed to protect the formed fibrin from becoming prematurely degraded, keeping the fibrinolytic activity somewhat delayed until the healing processes get started.
Structure
The gene of antiplasmin has been localized to chromosome 17p13.14 This inhibitor is a glycoprotein consisting of a single polypeptide, containing approximately 13% carbohydrate.15 Holmes et al.16 determined the primary structure of mature human α2-antiplasmin by DNA sequencing of overlapping cDNA fragments prepared from human liver mRNA. It was found to contain 452 amino acids as deduced from the cDNA sequence, with Asn as NH2-terminal amino acid. Later, it was found that native antiplasmin has an additional 12 amino acids at the NH2-terminal end and is rather composed of 464 amino acids with an NH2-terminal Met.17,18 In plasma, Metantiplasmin is converted to Asn-antiplasmin.19 There are four potential asparagine-linked glycosylation sites in the molecule. Antiplasmin has not yet been possible to crystallize, so much of its structure had to be anticipated by homologies with other proteins. It belongs to the serpin superfamily of proteins. It seems to contain all the important structural elements of a serpin. Of these, the reactive center loop (RCL), which contains the scissile peptide bond (red string in FIGURE 21.2) and the A β-sheet (yellow part in FIGURE 21.2), is of special functional importance. RCL is the part catching the protease and thus the most important portion of a serpin. It might occasionally be inserted into the A β-sheet as a new central strand and then form an inactive,
latent form that may be reactivated, if treated with denaturing substances, followed by dialysis. Alternatively, the RCL might be inserted into the A β-sheet of another molecule resulting in a tendency of polymerization.
latent form that may be reactivated, if treated with denaturing substances, followed by dialysis. Alternatively, the RCL might be inserted into the A β-sheet of another molecule resulting in a tendency of polymerization.
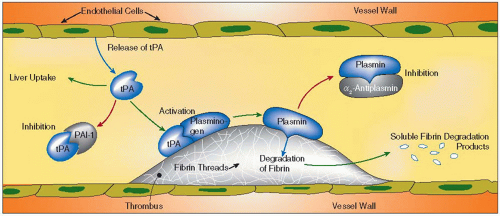 FIGURE 21.1 A schematic picture of the fibrinolytic system and its activation is demonstrated. Activation of plasminogen occurs preferably at the surface of fibrin, while plasmin becomes inhibited only after it has left the fibrin surface. (Adapted from Wiman, MFR informerar, 1987.) |
Reaction with Plasmin
Antiplasmin is capable of reacting with and forming stable enzymatically inactive complexes with plasmin. The reaction is very rapid, with a rate constant of about 2 × 107 M-1s-1, and occurs in several steps involving several different interacting sites.9,10,11,20,21 The first step, which seems to be the rate limiting, takes place between one of the lysine-binding sites in the plasmin molecule and a complementary site in the very COOHterminal (CT) portion of the antiplasmin molecule. The second is a noncovalent interaction between the substrate-binding pocket in the plasmin active site and the scissile peptide bond in the RCL of antiplasmin. Subsequently, peptide bond cleavage occurs.22 After formation of an ester bond between plasmin and antiplasmin, major conformational changes occur both in the plasmin molecule and in the antiplasmin molecule.
The lysine-binding-site-mediated interaction has an important role in regulating the fibrinolytic system. The same site in the plasmin molecules are also involved in the interaction with fibrin. Therefore, fibrin-bound plasmin reacts much slower with antiplasmin than free plasmin, thereby keeping the fibrinolytic process localized.2,9,11 It was initially believed that the CT lysine (Lys452) in antiplasmin is responsible for the lysine-binding-site-mediated interactions with different proteins.11,23,24,25,26,27,28,29,30,31 Current evidence indicates that K436, but not K452, is important in this reaction.32 Using single-site mutants, the variant K436E reacted much more slowly than wild-type antiplasmin (wt-antiplasmin) or the other variants including K452E or K452T. In addition, in the presence of 6-aminohexanoic acids, the reaction rate decreased about 10-fold or more for wt-antiplasmin and most variants. The variant K436E was less affected by 6-aminohexanoic acids, again suggesting that this residue is involved in the lysine-binding-site-mediated interaction between plasmin and antiplasmin. The data were further supported by studies using surface plasmon resonance to examine the interaction of antiplasmin mutants to immobilized plasminogen or an elastase fragment from plasminogen constituting “kringles” 1-3. The only antiplasmin variant with decreased affinity was the K436E, again demonstrating that the lysine residue 436 is important in this interaction.33 Thus, available data clearly demonstrate that Lys436 of antiplasmin is involved in the lysine-binding-site-mediated interaction with plasminogen and fibrin.
Stability
Many of the serpins seem to be metastable proteins, with quite interesting mechanisms regulating their stability. In contrast to some of the other serpins, human antiplasmin is quite stable at physiologic conditions, but that activity is rapidly lost at acidic conditions34 or at neutral pH at elevated temperatures.35 At temperatures above 45°C, the rapid inactivation was found to be a polymerization process. However, the inactivation occurring at low pH occurred mostly without any visible polymerization tendency. However, such material could gradually recover activity at neutral pH, and if treated with guanidinium chloride, followed by dialysis, antiplasmin activity was again recovered,35 in a similar fashion as for PAI-1 (see below).
Using site-directed mutagenesis, four single-site mutants, His197Thr, His341Thr, Glu353Ala, and Glu382His, were found to display significantly increased stability.36 The mutant His341Thr was found to have the longest half-life at pH 4.9, about sevenfold increased stability as compared to wt-antiplasmin. One mutant, Glu346Thr, had a shorter half-life
than wt-antiplasmin. All these residues are believed to be situated in the vicinity of the point where the RCL is inserted into the A β-sheet to get latent molecules (FIGURE 21.2). Unfortunately, so far it has not been possible to crystallize antiplasmin. However, the serpin superfamily of proteins all seem to be very similar in their overall structure. Therefore, in FIGURE 21.2, a schematic picture of PAI-1 is shown as adapted from Aertgeerts et al.37 1994. The expected places for where the important mutations in antiplasmin, as discussed above, would be situated are indicated in this figure.
than wt-antiplasmin. All these residues are believed to be situated in the vicinity of the point where the RCL is inserted into the A β-sheet to get latent molecules (FIGURE 21.2). Unfortunately, so far it has not been possible to crystallize antiplasmin. However, the serpin superfamily of proteins all seem to be very similar in their overall structure. Therefore, in FIGURE 21.2, a schematic picture of PAI-1 is shown as adapted from Aertgeerts et al.37 1994. The expected places for where the important mutations in antiplasmin, as discussed above, would be situated are indicated in this figure.
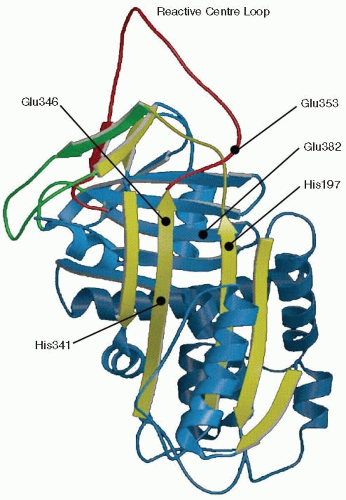 FIGURE 21.2 A schematic picture of active PAI-1 (Adapted from Aertgeerts et al. 1994). The expected sites for some mutations if this was in antiplasmin are indicated. Antiplasmin has not yet been crystallized. Therefore, we show a model of PAI-1 instead, since most serpins are expected to have a very similar basic structure. The RCL is labeled red and the A β-sheet yellow. |
Incorporation into Fibrin
Antiplasmin is incorporated into the α-chains of fibrin during the clotting process through the action of activated coagulation factor XIII. This interaction involves the NH2-terminal portion of antiplasmin, more specifically the Gln residue number 2. Only a fraction of the fibrin monomers will have an antiplasmin incorporated, but these molecules are believed to be very important from the physiologic point of view. They catch and inactivate the initial plasmin molecules formed upon activation of the fibrinolytic system. In that way, fibrin is initially protected and the fibrinolytic process becomes somewhat delayed.38 This permits the repair processes to get started before the fibrinolytic process really gets accelerated.
Clinical Significance
A few individuals have been identified with congenital antiplasmin deficiency.39 They suffer from a severe bleeding tendency, similar to individuals with classical hemophilia. However, when correctly diagnosed, these patients are very easily treated with an tifibrinolytic drugs. Tranexamic acid or earlier 6-aminohexanoic acid is the most frequently used drug to achieve this. It interferes with the interaction between plasminogen and fibrin and prevents formation of the ternary complex between fibrin, plasminogen, and tPA (tissue plasminogen activator). This causes a dramatic decrease in the activation rate of plasminogen and thus, an antifibrinolytic effect is obtained. Aprotinin, a low molecular weight and very efficient inhibitor of trypsin and plasmin, has also been used. Decreased plasma concentrations of antiplasmin are seen in connection with severe liver disease.
PAI-1
Discovery and Basic Information
PAI-1 is a serpin with a molecular weight of about 50,000. It was initially discovered in endothelial cells40 in human plasma41,42,43 and in hepatocytes.44 It is now known that PAI-1 is produced by several types of cells, including vascular endothelial cells, adipocytes, platelets (or rather megakaryocytes), and placental cells.1 PAI-1 is also produced by smooth muscle cells in atherosclerotic arteries.1
The concentration of active PAI-1 in human plasma is 0.6 nmol/L (about 30 µg/L), much lower than for the other serpins. Some individuals have 5 to 10-fold higher plasma concentrations. This is true for individuals with increased body weight, especially those with an increased waist/hip ratio, or those with an increased inflammation, but it is also true for pregnant women.
PAI-1 efficiently inhibits tPA and uPA (urinary plasminogen activator) by forming a 1:1 very stable complex that resists strong denaturing substances such as SDS.45 The reaction between PAI-1 and tPA/uPA is also extremely fast with second-order rate constants above 107 M-1 s-1.46,47 During complex formation, the scissile peptide bond Met346-Arg347 in PAI-1 becomes cleaved.45,48
The PAI-1 Gene
The human PAI-1 gene is located on chromosome 7 and is approximately 12.2 kbp long. The gene contains nine exons separated by eight introns. Several groups have sequenced and analyzed this gene.49,50,51,52,53,54,55 Two different PAI-1 mRNA species seem to exist in humans with lengths of 2.4 and 3.2 kb.50 They both contain the entire coding region and just differ in the length in their 3′-untranslated region.51,56 Both these messengers seem to function similarly, but they differ in stability, with the 2.4-kb form being more stable.57 The different transcripts are the result of the presence of multiple polyadenylation splice sites.58
Several polymorphisms within the PAI-1 gene have been described. The most frequently studied one is the 4G/5G polymorphism located 675 bp upstream of the transcriptional start site.59,60 It is a polymorphism of functional importance since it correlates with the plasma PAI-1 concentration (see below). Other reported polymorphisms are a HindIII polymorphism in the 3′-region,56 a (CA)n repeat in intron 3,60 a G/A substitution at -844 in the promoter61,62 and at +1334194, a T/G substitution at +11053,62 a 9-nucleotide insertion/deletion between +11320 and 11345,62 and a G/A substitution at position +9785 in intron 7.62 None of these have yet been shown to be of any practical importance.
Structure
Human PAI-1 is a single-chain glycoprotein with a molecular weight of about 50,000.42 Recombinant PAI-1 expressed in Escherichia coli has a molecular weight of approximately 43,000 due to the lack of glycosylation in the bacteria. After removal of a signal peptide of 23 amino acids, the mature protein consists of 379 amino acids. PAI-1 lacks cysteine residues and contains three potential glycosylation sites.50 It has the typical serpin structure (FIGURE 21.2).
The PAI-1 molecule is fully active upon synthesis,63 but under physiologic conditions it seems to be spontaneously transformed into an inactive form. This form can, however, again become active if treated with some denaturing (preferably hydrogen bond-cleaving) substances and allowed to refold.40,63,64 This form has come to be called “latent” PAI-1. The crystal structure of latent PAI-1 was solved in 199265 and revealed that in the latent conformation the RCL (red string in FIGURE 21.2) is fully inserted forming a new strand in the middle of the A β-sheet37 (yellow portion in FIGURE 21.2). In the active conformation, the RCL is exposed on the surface of the molecule (see FIGURE 21.2).
The Reaction between PAI-1 and Plasminogen Activators
As mentioned above, the reactions between PAI-1 and the different plasminogen activators, tPA or uPA, are very rapid. This seems to depend on the presence of second-site interactions46 in analogy with the reaction between plasmin and antiplasmin.9,20 Besides the interaction between the active site in the activators and the scissile peptide bond Met346-Arg347 in the RCL in PAI-1, an interaction between Glu350 in PAI-1 with Arg304 in tPA also seems to be of importance.66,67
The formation of the stable complex between a serpin and a serine proteinase occurs by the binding of the RCL to the active site of the proteinase, followed by cleavage of the reactive center peptide bond and formation of an ester bond between the carboxyl group of the P1 residue of the serpin and the hydroxyl group of the active-site serine of the proteinase (P1P1′).22,48,68,69 The amino acid residues at positions P1-P16 are then inserted into the A β-sheet, and the serine proteinase is transported toward the opposite pole of the RCL in the serpin.48,69,70,71
The inhibition of plasminogen activators by PAI-1 involves an interaction between the RCL of the inhibitor and the catalytic groove of the proteinase in question. The mobility of the RCL is required for the inhibitory function of PAI-1 but not for the initial recognition of the plasminogen activators.72 The basic residue at the P1 position within the RCL plays a crucial role for the uPA inhibitory activity.73,74,75 However, the presence of a neutral or hydrophobic residue at this position does not affect the inhibitory activity toward tPA.76
Different Molecular Forms of PAI-1 and Stability
The existence of latent PAI-1 was described above. The transformation of active to latent inhibitor is a spontaneous process at physiologic conditions. The importance of some residues on β-strand 5A for the activity and latency transition of PAI-1 has been investigated.77 It was shown that the side chain of Gln303 is important since substitution of this residue toward a His increased the latency transition fourfold. Binding of a murine monoclonal antibody produced against the human tPA-PAI-1 complex induced a 4,000-fold decrease in the half-life of active PAI-1.78 Binding of this antibody seems to cause a partial insertion of the RCL into the PAI-1 A β-sheet. Different parts of helix F in the PAI-1 molecule seem also to be involved in the conversion from active to the latent form. When substituting Asp125 by Lys and Arg133 by Asp, these variants were rapidly transformed into latent forms.79 The residue Lys325, situated at β-strand s5A and normally forming a hydrogen bond with Asn150 (located on the s3A/hF loop), also seems to be important in regulating the latency transition.80
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Molecular Basis for Platelet Secretion
Molecular Basis for Platelet Secretion
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


