BSS: First reported in 1948, BSS associates macrothrombocytopenia with decreased platelet adhesion to subendotheliumcausedby an absence (or nonfunctioning) of GPIbα3,4,5 A simple diagnostic test is that the giant platelets fail to agglutinate with ristocetin, an antibiotic that promotes the binding of von Willebrand factor (vWF) to GPIbα. The products of four separate genes (GPIBA, GPIBB, GP9, and GP5) assemble in a 2:2:2:1 ratio within maturing MK to form the GPIb-IX-V complex, stabilized through cytoplasmic
domains linked to the membrane cytoskeleton via filamin A (FlnA).6 Generally inherited as an autosomal recessive trait, mutations within GPIBA, GPIBB, and GP9 interfere with the biosynthesis, maturation, and/or trafficking of the complex through the endoplasmic reticulum and Golgi apparatus of MKs.7,8 Recent data suggest that bleeding results from the loss of high shear- and vWFdependent platelet-to-platelet interactions in thrombus formation, as well as a defective platelet adhesion and the low platelet count.9 The additional absence of GPIbαdependent binding of P-selectin, TSP1, factor VIIa, factor XI, factor XII, aMβ2, high molecular weight kininogen, and thrombin may also contribute to the phenotype (see Chapter 26A). Early reports of an increased phosphatidylserine (PS) exposure but decreased prothrombin consumption and a decreased platelet survival in BSS remain largely unexplained. Nonetheless, BSS platelets of all sizes readily expose PS and undergo apoptotic-like changes on platelet activation.10
Early studies showed that mature MKs isolated from the bone marrow of BSS patients had major changes in morphology with an altered organization of internal membranes, many vacuoles, and a disparate localization of α-granules.11 Such changes lead to decreased platelet production with the formation of large round platelets having distinctive morphologic abnormalities (FIGURE 63.1). The macrothrombocytopenia in BSS is usually moderate but can be severe in some patients and lead to extensive, occasionally life-threatening, mucocutaneous bleeding. In rare variant forms, platelets express decreased to normal amounts of nonfunctional GPIbα. Mutations resulting in variant BSS have included three missense mutations in GPIbα: R41H, L73F, and A156V (so-called Bolzano mutation).12,13,14 While the first two were heterozygous with autosomal dominant pattern of inheritance, the Bolzano variant was homozygous with autosomal recessive inheritance. Hemizygous mutations in GPIBB can also cause BSS when associated with the DiGeorge/velocardiofacial syndrome, a developmental disorder characterized by a hemizygous microdeletion at 22q11, the site of localization of GPIBB.15
Mice lacking GPIbα or GPIbβ have allowed evaluation of megakaryocytopoiesis in BSS and have confirmed aberrant membrane development early during MK maturation leading to a reduced demarcation membrane system (DMS), impaired proplatelet formation, and aberrant microtubule coil assembly.16,17,18 Studies of platelet production in situ revealed large platelets being released from unusually short, broad, proplatelets protruding into vascular sinuses.17
GPIbβ—/— MKs differentiated in vitro showed fewer proplatelet extensions with thicker shafts, fewer branches, and an increased diameter of terminal coiled elements that were enriched in tubulin fibers.18 Significantly, the macrothrombocytopenia was much less severe in a novel murine model where platelets were engineered to express a GP1393;bα subunit in which the extracytoplasmic sequence was replaced by that of the α-subunit of the human interleukin-4 receptor; despite improvement in platelet production a severe bleeding phenotype persisted.19,20 This suggests a role for the GP1393;bα cytoplasmic tail in signaling essential for normal MK maturation. Significantly, no correlations have yet been seen in BSS between phenotype (and bleeding severity) and genotype.21
Mediterranean Macrothrombocytopenia. In Mediterranean countries, patients manifesting a moderately low platelet count (70,000 to 150,000/µL), increased mean platelet volume, autosomal dominant inheritance with mild mucocutaneous or no bleeding, normal platelet function, and normal MK numbers are frequently observed. Linkage analysis and mutation screening of 12 Italian families identified a heterozygous A156 substitution in GPIbα in six with GPIb-IX levels comparable to those of BSS heterozygotes.22 It is noteworthy that A156V is the same mutation as that causing the Bolzano variant of BSS (see above). Balduini et al.23 cultured MKs from six patients and showed that proplatelet formation was much reduced compared to controls. Again, proplatelet tips were enlarged and α-tubulin distribution abnormal. Because the GPIbα mutation is heterozygous and occurs in the context of the wild-type protein, it must be exerting an as yet unexplained dominant negative effect, especially since obligate heterozygotes for classic BSS are largely asymptomatic.
Platelet-type von Willebrand disease (platelet-type vWD). Platelet-type vWD is characterized by a gain of function phenotype with spontaneous binding of plasma vWF to platelets and increased platelet agglutination by low amounts of ristocetin in the presence of normal plasma. Platelet size can be increased in platelet-type vWD and there can be moderate thrombocytopenia. Selected heterozygous GPIbα substitutions with autosomal dominant inheritance cause platelet-type vWD, namely, G233V/S and M239V substitutions in the N-terminal domain; a 27-bp deletion in the macrogly copeptide-coding region of GPIBA implies that long-range conformational changes can also give rise to platelet-type vWD.4,24,25 Mechanistically, the GPIbα mutations promote and stabilize platelet adhesion to vWF at low shear by enhancing formation and increasing longevity of the tether bond.26 Interestingly, a knock-in mouse model with the G233V mutation not only had a phenotype mimicking the human disorder, but also showed increased bone mass.27 In the same model, spontaneous binding of vWF blocked the capacity of platelets to bind fibrinogen when stimulated.28 This probably contributes to the bleeding caused by an inhibited GPIbα although enhanced ADAMTS13 cleavage of the more stable plateletbound vWF multimers may moderate the condition.29
vWD2B. Enlarged platelets with size heterogeneity (FIGURE 63.1) and thrombocytopenia can also occur in vWD, type 2B (vWD2B) where mutated plasma vWF spontaneously binds to a normal GPIbα.30 As with platelet-type vWD, inheritance is autosomal dominant. In one such family with an R1308P vWF substitution, platelets showed signs of apoptosis and culture of CD34+ cells from the peripheral blood resulted in an increased surface expression of the mutant vWF on mature MKs that caused interactions between proplatelets.31 This study was extended to nine patients with a total of seven different gain-offunction mutations and abnormal platelets typical of those seen in the circulation were produced ex vivo in MK cultures.32 Circulating platelet clumps in rare patients with vWD2B prompted comparison with the previously described phenotype of the Montreal platelet syndrome (MPS); this was resolved when patients of the founder MPS kindred were shown to have the common vWF V1316M mutation.33 Among the described characteristics of MPS were platelet calpain deficiency (probably due to autodegradation) and a reduced thrombin-induced aggregation; both findings are a consequence of the spontaneous binding of large vWF multimers to GPIbα. Significantly, lineage-specific mouse knock-in models of vWD2B show that abnormal plasma vWF (with a vWD2B mutation) can broadly reproduce the human vWD2B phenotype, emphasize its hemostatic variability, and suggest a possible role for ADAMTS13-dependent modulation of disease severity.34,35
Table 63.1 Inherited thrombocytopenias classified by gene mutation and associated phenotype | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
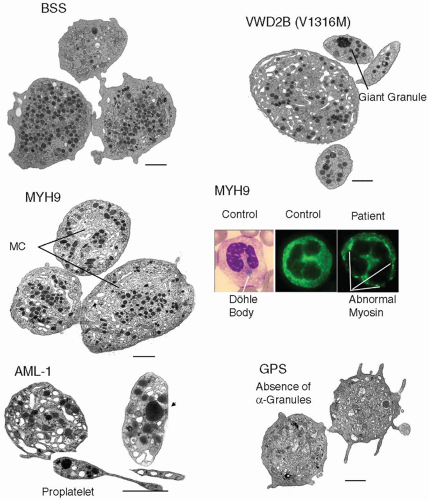 FIGURE 63.1 Composite illustration showing some of the striking ultrastructural characteristics of platelets in selected inherited thrombocytopenias. In the upper panels are shown the typical large round platelets of a patient with the BSS as seen by electron microscopy. Note the heterogeneous distribution of α-granules but the absence of large MC. Adjacent are platelets from a vWD2B patient showing size heterogeneity. An enlarged α-granule is highlighted. The middle panels first show electron microscope images of large round platelets from a patient with MYH9-related disease (May-Hegglin anomaly); note the large abnormal platelets with abundant MC. Also shown are a cytochemically stained Döhle-like inclusion and NMMHC-IIA immunoprecipitates detected by immunofluorescence in leukocytes from the same patient. In the lower panel is first shown platelet anisotropy in a patient with amegakaryocytic thrombocytopenia with predisposition to leukemia (AML-1). Platelets are immature and proplatelet fragments are present. Finally two platelets from a patient with the GPS are shown. These platelets are totally lacking α-granules. Bars = 1 µm |
Döhle-like inclusions in neutrophils, eosinophils, and some monocytes are diagnostic characteristics (FIGURE 63.1).45 Thrombocytopenia is generally mild but can be severe; bleeding is infrequent and rarely life threatening. Other manifestations of these disorders include the progressive development of nephritis, sensorineural hearing loss, and cataracts. The Sebastian platelet syndrome associates macrothrombocytopenia with neutrophilic inclusions that differ in morphology from those in May Hegglin anomaly.46 The Fechtner syndrome adds hearing loss, nephritis, and cataracts while in the Epstein syndrome only hearing loss and glomerulonephritis are present (Table 63.1).47,48
Mutation of the β1-tubulin gene (TUBB1). Microtubule assembly and function are key elements of proplatelet formation from MKs, trafficking granules and other organelles to the proplatelet tips where platelets are released by budding, and maintenance of the platelet discoid shape.71,72 Freson et al.73 found a heterozygous Q43P polymorphism in a highly conserved residue of β1-tubulin in 24.2% of 33 unrelated families with inherited macrothrombocytopenia of unknown cause. Electron microscopy showed enlarged spherocytic platelets with a disturbed marginal microtubule band and organelle-free zones. Decreased platelet reactivity to physiologic agonists was also noted. However the same mutation was also seen in approximately 10% of normal subjects. Kunishima et al.74 studied a Japanese patient with the heterozygous Q43P mutation but found that it did not cause macrothrombocytopenia. Instead, these authors found a second heterozygous R318W mutation predicted to disrupt side chain interactions near the α and β intradimer interface of β1 -tubulin. The authors found that R318W, but not Q43P, led to a 50% reduction in platelet β1-tubulin content. Cultured MKs from the patient showed morphologic changes consistent with a disturbed platelet production; the R318W β-tubulin was localized in punctate structures suggesting that it was forming aggregates. This unstable protein may be essential for the phenotype; while β1-tubulin (-/-) mice are thrombocytopenic and their platelets lose their discoid shape, heterozygotes do not show macrothrombocytopenia.75
Mutations in the filamin A gene (FLNA). Heterozygous mutations in the FLNA gene encoding the cytoskeletal protein FlnA are associated with a series of rare X-linked autosomal dominant diseases, the major feature being periventricular nodular heterotopia (PNH).76 FlnA is the attachment site for GP1393;bα in the platelet cytoskeleton, helping to maintain thrombus stability at high shear.77 Bleeding is an important feature for a cohort of patients with FLNA mutations as is thrombocytopenia and platelet anisotropy.78 This was shown to be due to a defective megakaryocytopoiesis, a finding confirmed from studies on a conditional mouse model where FlnA null platelets also showed signaling defects and a reduced shear-dependent platelet accumulation on collagen.79 Mice whose platelets possessed intermediate amounts of FlnA also showed macrothrombocytopenia.Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


