hemorrhage. Hemarthroses and expanding hematomas are unusual. Although the severity of bleeding is variable and may not always correlate with the extent of thrombocytopenia and the degree of GPIb-IX-V deficiency,35 it can be severe enough to require transfusion and suppression of menses. In a review of 59 patients, for example, there were 10 deaths.36 In some patients with BSS, the severity of hemorrhage inexplicably declines over the course of the disease.36
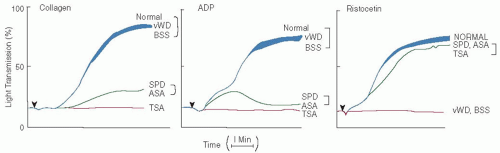 FIGURE 65.1 Platelet aggregation in various platelet disorders. Platelet aggregation can be studied ex vivo by a variety of techniques. Conventional turbidometric platelet aggregometry is performed in an “aggregometer” using stirred suspensions of either unwashed platelets in plasma or washed platelets in buffer. Immediately after the addition of all platelet agonists except epinephrine, there is a transient decrease in light transmittance that has been attributed to platelet shape change. This is followed by an increase in light transmission as platelet aggregates form. Two “waves” of platelet aggregation have been described. The “first wave” represents primary aggregation and is a direct consequence of agonist stimulation. The “second wave” represents secondary aggregation and is due to the formation of large irreversible aggregates when platelet secretion occurs. If the concentration of platelet agonist is sufficiently high, the first and second waves merge into a single continuous tracing. Measurements of platelet aggregation are expressed as either the extent or the rate of increase in light transmittance that occurs as platelet aggregates form. The turbidometric aggregometer has been modified to simultaneously measure platelet aggregation and platelet dense granule secretion using the luciferin-luciferase reaction to measure secretion of dense granule ATP (lumiaggregometry). Platelet aggregation can also be measured in diluted anticoagulated whole blood based on the increase in electrical impedance (measured in ohms) that occurs as platelet aggregates accumulate on a pair of electrodes inserted into the suspension of aggregating platelets (impedance aggregometry). Impedance aggregometry tracings are similar to those obtained by the turbidometric method, except that shape change and two waves of aggregation are not seen. Lastly, because aggregometry is insensitive to the formation of small platelet aggregates, methods have been devised to measure the disappearance of single platelets immediately after platelet stimulation. Agonists are added to stirred platelet suspensions and after adding a fixative to stabilize the aggregates, their number and size are measured either visually with a hemocytometer or using an electronic particle counter or flow cytometer. The platelet aggregation tracings shown in the figure were generated in a conventional turbidometric platelet aggregometer after the addition of collagen, ADP, and ristocetin to stirred aliquots of platelet-rich plasma. vWD, von Willebrand disease; BSS, Bernard-Soulier syndrome; SPD, storage pool disease; ASA, aspirin; TSA, Glanzmann thrombasthenia. (Adapted from Weiss HJ. Platelet physiology and abnormalities of platelet function (second of two parts). N Engl J Med 1975;293:580-588, with permission.) |
Although the serum prothrombin time has been reported to be decreased in BSS47 and both collagen-induced platelet procoagulant activity and factor XI binding to be absent,48 prothrombin consumption has also been reported to be normal49 and platelet procoagulant49 and prothrombinase activity to be increased.50 An unexplained decrease in thrombin-, trypsin-, and thromboxane-stimulated phospholipase C (PLC) activity has also been reported.51 GPIb-IX-V can be a target for drug-dependent anti-platelet antibodies52; such antibodies fail to bind to BSS platelets.53
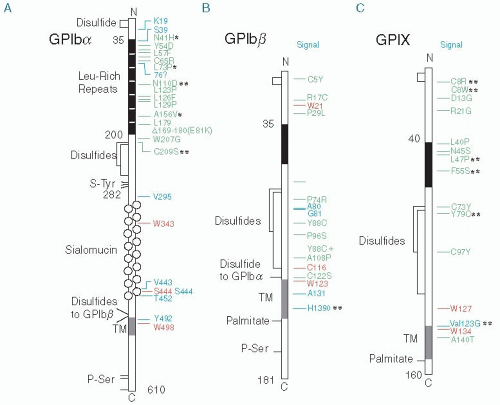 FIGURE 65.2 Mutations in the genes for GPIbα, GPIbβ, and GPIX causing the BSS mapped onto the structures of the mature proteins. Green, missense mutations and short deletions; red, nonsense mutations leading to premature stop; and blue, frameshift mutations leading to stop. *, autosomal dominant inheritance; **, mutations discussed in Savoia A, Pastore A, de Rocco D, et al. Clinical and genetic aspects of Bernard-Soulier syndrome: searching for genotype/phenotype correlations. Haematologica 2011;96:417-423. (Adapted from Berndt MC, Andrews RK. Bernard-Soulier syndrome. Haematologica 2011;96:355-359, with permission.) |
are involved in efficient GPIb-IX-V expression69 and with the impairment of GPIb-IX expression observed when the GPIbβ transmembrane domain was mutated in vitro.70 The monoallelic GPIbα mutation Ala156Val, known as the “Bolzano mutation,” has been reported to be the most common cause of inherited thrombocytopenia in Italy. It has been associated with a mild bleeding diathesis, a variable degree of thrombocytopenia, and a modest but consistent increase in platelet diameter and volume.71 It is also noteworthy that many BSS mutations have been reported multiple times, suggesting that there may be “hot spots” for mutation in the GPIbα, GPIbβ, and GPIX genes.
Table 65.1 Differential diagnosis of inherited macrothrombocytopenia | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
to type 2B vWD caused by a heterozygous V1316M mutation in exon 28 of the vWF gene.85,86 Mediterranean macrothrombocytopenia refers to putative differences in platelet count and size between Europeans of Northern and Mediterranean origin87; in Italy, this may be due to heterozygosity for BSS, in particular heterozygosity for the “Bolzano mutation” Ala156Val.88 Mediterranean stomatocytosis/macrothrombocytopenia, the rare combination of stomatocytic hemolysis and macrothrombocytopenia, results from excess plant sterols in the blood and is caused by mutations in the ABCG5 and ABCG8 genes linked to phytosterolemia.89 Several reported patients had significant bleeding histories. Sex-linked macrothrombocytopenia occurs in patients with GATA-1 transcription factor mutations,90,91,92 and macrothrombocytopenia with dystrophic megakaryocytes and neutrophils lacking sialyl-Lewis-S antigen was described in an infant who died at 37 months due to hemorrhage.93 In the Paris-Trousseau syndrome,94 a part of the Jacobsen syndrome,95 a deletion of the long arm of chromosome 11 at q23.3 containing the gene for the transcription factor FLI1 results in mild thrombocytopenia, subpopulations of large platelets and platelets containing giant α-granules, and the expansion of immature megakaryocyte progenitors.96 Mutations in the filamin A gene located on chromosome Xq28 are responsible for periventricular nodular heterotopia and the otopalatodigital syndrome; macrothrombocytopenia and bleeding, misinterpreted as ITP, has been present in some patients.97 In platelet-type vWD, mutations within the vWF binding domain of GPIbα induce spontaneous vWF binding to GPIbα and the clinical picture of type IIB vWD which itself can be associated with macrothrombocytopenia.9,98,99,100,101,102 There are also anecdotal reports of thrombocytopenia and giant platelets, often associated with a mild bleeding diathesis but lacking the features of either BSS or the MHY9 disorders.103,104 In two other reports, macrothrombocytopenia was associated with the presence of large platelet granules whose origin was uncertain.105,106
(GT) results when platelets lack sufficient numbers of functional αIIbβ3 to support platelet aggregation.128 However, the platelets of obligate thrombasthenic heterozygotes aggregate normally, indicating that 50% of the normal amount of αIIbβ3 is at least sufficient to support platelet aggregation.147
Similarly, an insertion of two amino acids (Arg-Thr) into the Cys146-Cys167 loop of αIIb produced an inactive αIIbβ3 heterodimer.190 An eighth mutation, β3 Ser123Pro, did not impair αIIbβ3 expression or agonist-induced ligand binding but was associated with absent ADP- and collagen-induced platelet aggregation.191 The β3 mutation, Leu196Pro, was identified independently in two French patients and severely restricted αIIbβ3 expression.192,193 However, the residual αIIbβ3 that made it to the platelet surface was unable to bind ligands, presumably because the mutation prevents αIIbβ3 from assuming its active conformation. By contract, the β3 mutation Cys560Arg locked αIIbβ3 in a high-affinity conformation, but like Leu196Pro, it likely produced a GT-phenotype because the small amount of constitutively active αIIbβ3 on the platelet surface was insufficient to support platelet aggregation.194
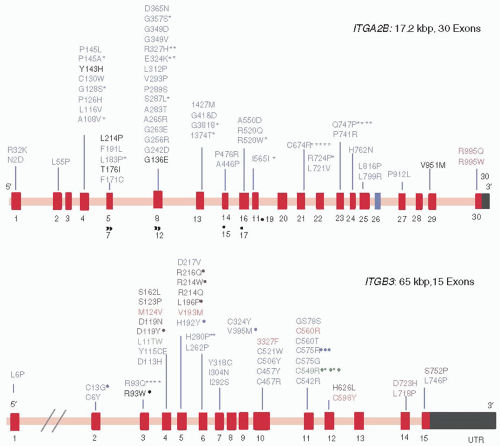 FIGURE 65.3 Missense mutations in the ITGA2B and ITGB3 genes resulting in GT. The exons in the ITGA2B and ITGB3 genes are shown schematically as red bars. Mutations shown in black are lack of function mutations, those shown in blue primarily impair αIIbβ3 expression, those in green are characteristic of particular ethnic groups, those in orange cause constitutive αIIbβ3 activation, and those in rose are associated with macrothrombocytopenia. Asterisk indicate the number of times the mutation has been reported. (Adapted from Nurden AT, Fiore M, Nurden P, et al. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood 2011;118:5996-6005, with permission.) |
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


