Introduction
Non-Hodgkin B-cell lymphomas are the seventh most common malignant neoplasm and account for approximately 4% of all cancers, according to the American Cancer Society, with approximately 77,000 cases anticipated to be diagnosed annually in the United States, not including an additional 21,000 estimated cases of chronic lymphocytic leukemia ( https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2020.html ). B-cell neoplasms often mimic normal stages of B-cell maturation from lymphoblasts generated in the bone marrow to a spectrum of mature peripheral B cells, including naive B, germinal center, and memory B cells and plasma cells. Association with particular B-cell stages is more than a diagnostic aid; it may also have functional consequences for the pattern of tumor progression. For example, somatic hypermutation of IGV and BCL6 occur in the germinal center, and germinal center B-cell–derived neoplasms have ongoing IGV mutation and greater intraclonal diversity. By contrast, post-germinal center neoplasms such as marginal zone lymphomas have mutated IGV but no evidence of ongoing mutation. Different lymphomas also demonstrate different predilections for various anatomic sites. This chapter flows from more indolent to more aggressive disorders in addition to highlighting the most common disease entities first. Within each disease entity, however, there may be a range of indolent to more aggressive forms. In particular, follicular lymphoma and mantle cell lymphoma (also highlighted in Tables 12.1 and 12.2 ) have distinct subtypes with important prognostic and therapeutic implications. There have been tremendous advances in the past 5 years in the understanding of the genetic landscape of B-cell lymphomas. This topic is too broad to cover in this chapter, but highlights are featured.
| Demographics | Clinical Course | Salient Features | |
|---|---|---|---|
| Follicular lymphoma (usual type) | Median age 59 years | Dependent on grade | Follicular structure, 17% with grade 3A or 3B morphology CD10 + MUM1 − , BCL2 + , BCL6 + IGH/BCL2 translocation present High rate of somatic mutation |
| In situ follicular neoplasia | Often associated with prior or concurrent lymphoma | Low rate of progression; may be detected by flow cytometry in 50% of cases | Must distinguish from partial nodal involvement by FL, may precede overt FL but not predicted by extent of involvement |
| Pediatric-type follicular lymphoma | Predominantly males, median age 15, predilection for head and neck | Low clinical stage, good prognosis | Follicular, cells with blastoid morphology, cannot have diffuse foci , No BCL2 , BCL6 , IRF4 , or aberrant IG rearrangment, no BCL2 amplification BCL6 positive, BCL2 negative or weakly positive, high KI67 proliferation rate |
| Duodenal-type follicular lymphoma | Localized to duodenum | Distinct from other FL of gastrointestinal tract, excellent prognosis | Follicular, low-grade, some features overlap with in situ follicular neoplasia, also overlap with extranodal marginal zone lymphoma |
| Diffuse-appearing follicular lymphoma | Often presents as large localized inguinal masses | Low clinical stage | Diffuse pattern but low-grade, residual germinal centers CD10 + , Bcl6 + , frequently CD23 +85 1p36 deletions, including TNFRSF14 or presence of TNFRSF14 mutations, STAT6 often mutated, lack BCL2 rearrangements |
| Large B-cell lymphoma with IRF4 rearrangement | Children and young adults, often Waldeyer ring or cervical nodes | Low clinical stage, more aggressive than pediatric-type FL, good prognosis when treated | Follicular, follicular and diffuse, or diffuse pattern; high proliferative index Strong IRF4/MUM1 expression, usually BCL6 + , BCL2 + and CD10 + in >50%, minority CD5 +84 IRF4 rearrangement (most), some also have BCL6 rearrangement, lack BCL2 rearrangement |
| CD10 − , IRF4/MUM1 + follicular lymphoma | Older individuals (median 67 years) | Often associated with DLBCL | Follicular structure, 91% with high-grade morphology Lack the BCL2/IGH translocation, most with BCL6 translocation or amplification High rate of somatic hypermutation |
| Clinical Course | Salient Features | |
|---|---|---|
| Mantle cell lymphoma (classic type) | Aggressive, typically involves lymph nodes and extranodal sites | SOX11 + , cyclin D1 + IGHV unmutated or minimally mutated If low proliferative fraction, may be more indolent |
| Blastoid or pleomorphic | More aggressive, may represent progression | Acquisition of additional molecular/cytogenetic abnormalities |
| Leukemic nonnodal mantle cell lymphoma | Frequently clinically indolent, usually involving peripheral blood, bone marrow, often spleen | SOX11 − IGHV mutated Secondary mutations, particularly involving TP53 , may result in very aggressive disease |
| In situ mantle cell neoplasia | Often an incidental finding, appears to have a low rate of progression, may be disseminated | Cyclin D1 + in inner mantle zones of follicles, in node without morphologic features of MCL; some SOX11 + (44%) 87 ; t(11;14) present Must distinguish from overt MCL with a mantle zone growth pattern |
Follicular Lymphoma
Follicular lymphoma accounts for approximately 40% of non-Hodgkin lymphoma (NHL) cases, with the highest rates in the United States, Western Europe, and Cape Town, South Africa. , It is predominantly seen in adults and is rare in patients younger than 20 years. Recent updates to the World Health Organization (WHO) classification have emphasized several subtypes of diagnostic and prognostic significance, including in situ follicular neoplasia (previously referred to as in situ follicular lymphoma) and pediatric-type, duodenal-type, and diffuse-appearing follicular lymphoma (see Table 12.1 ). ,
Follicular lymphoma is derived from germinal center B cells (centrocytes and centroblasts) where the germinal center cells fail to undergo apoptosis owing to the overexpression of BCL2, generally owing to an IGH/BCL2 t(14;18) translocation (present in 90% of cases). However, there have been recent advances in uncovering additional aspects of the mutational landscape important for the development and progression of this disorder ( Table 12.3 ).
| Cytogenetics | Characteristic Genetic Features | Prognostic Markers | |
|---|---|---|---|
| Follicular lymphoma | t(14;18) IGH-BCL2 | High rate of somatic mutation Chromatin modifiers: CREBBP, KMT2D ( MLL2 ), and EZH2 | TP53 Additional BCL2 mutations associated with increased risk of transformation |
| MALT lymphoma | Trisomies 3 and 18, t(11;18)(q21;q21), t(1;14)(p22;q32), t(14;18)(q32;q21), t(3;14)(p14.1;q32), loss of 6q23-24, del 7q31 | Biased use of certain IGHV families TNFAIP3 (6q23-24, 15%–30%) MYD88 L265P (6%–9%) | t(11;18)(q21;q21) typically resistant to antibiotics for Helicobacter pylori |
| Nodal MZL | Trisomies 3 and 18, loss of 6q23-24 | Favored IGHV3 , IGHV4 , particularly IGHV4-34 | |
| Splenic MZL | Trisomies 3 and 18, loss of 6q23-24 | ||
| Lymphoplasmacytic lymphoma | No specific | MYD88 L265P , CXCR4 | CXCR4 mutation may correlate with decreased response to ibrutinib |
| Mantle cell lymphoma | t(11;14)(q13;q32) (>95% of cases); CCND2 translocations with IGL or IGK | CCND1 (additional mutations; ATM, KMT2D ( MLL ); NOTCH1 and NOTCH2 , cell cycle regulators RB, p53, and CDK inhibitors all associated with an aggressive course | Assessment of proliferation rate is critical (>10 mitoses per 15 high-power fields or >30% Ki67 proliferation rate) associated with adverse prognosis |
Follicular lymphoma is composed of varying proportions of follicle center–type cells including centrocytes and centroblasts. Histologic grading according to WHO classification is determined by the number of centroblasts per high-power field, which represent the proliferative component. All low-grade follicular lymphoma subtypes where centrocytes predominate are combined into a single category of grade 1-2 ( Figs. 12.1 and 12.2 ), with fewer more than 15 centroblasts per high-power field. Grade 3 follicular lymphoma demonstrates more than 15 centroblasts per high-power field and should be further subdivided into 3A and 3B based on the presence or absence of background centrocytes. Grade 3B lacks background centrocytes and appears biologically to be more closely related to diffuse large B-cell lymphoma (DLBCL).

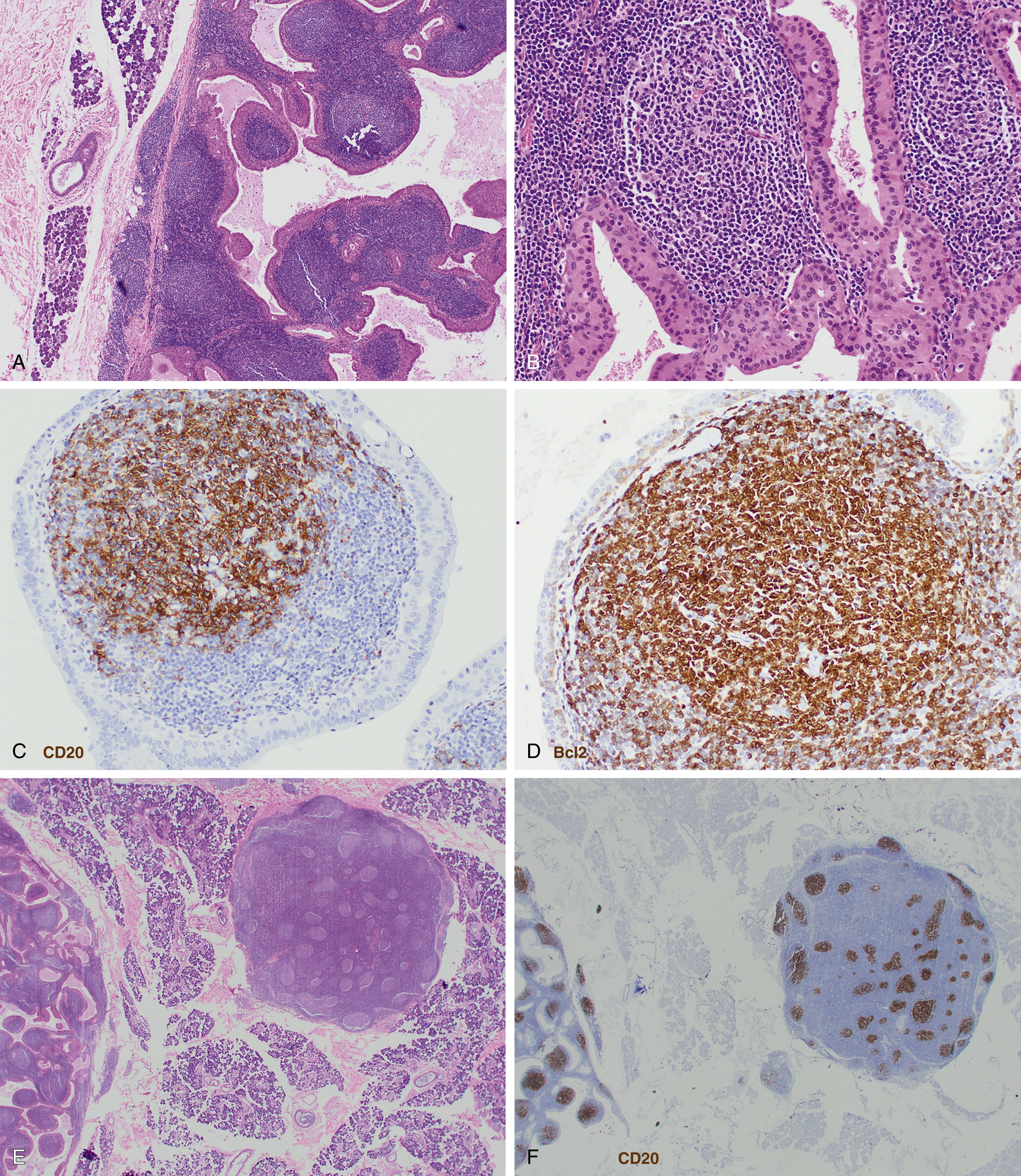
Clinical staging uses the Lugano classification, based on consortium recommendations for both Hodgkin and NHL. For follicular lymphoma, this includes an assessment of bone marrow involvement by biopsy.
In Situ Follicular Neoplasia
The revised WHO 2016 classification recognizes in situ follicular neoplasia as a distinct lesion. This is separate from partial involvement by follicular lymphoma. In situ follicular neoplasia is characterized by involvement of germinal centers by CD10 and BCL2-positive cells carrying a t(14;18) translocation in an otherwise reactive-appearing lymph node and is often detected as an incidental finding. Patients have a low rate of progression and a low risk of developing follicular lymphoma if not detected at the time of initial staging.
Pediatric-Type Follicular Lymphoma
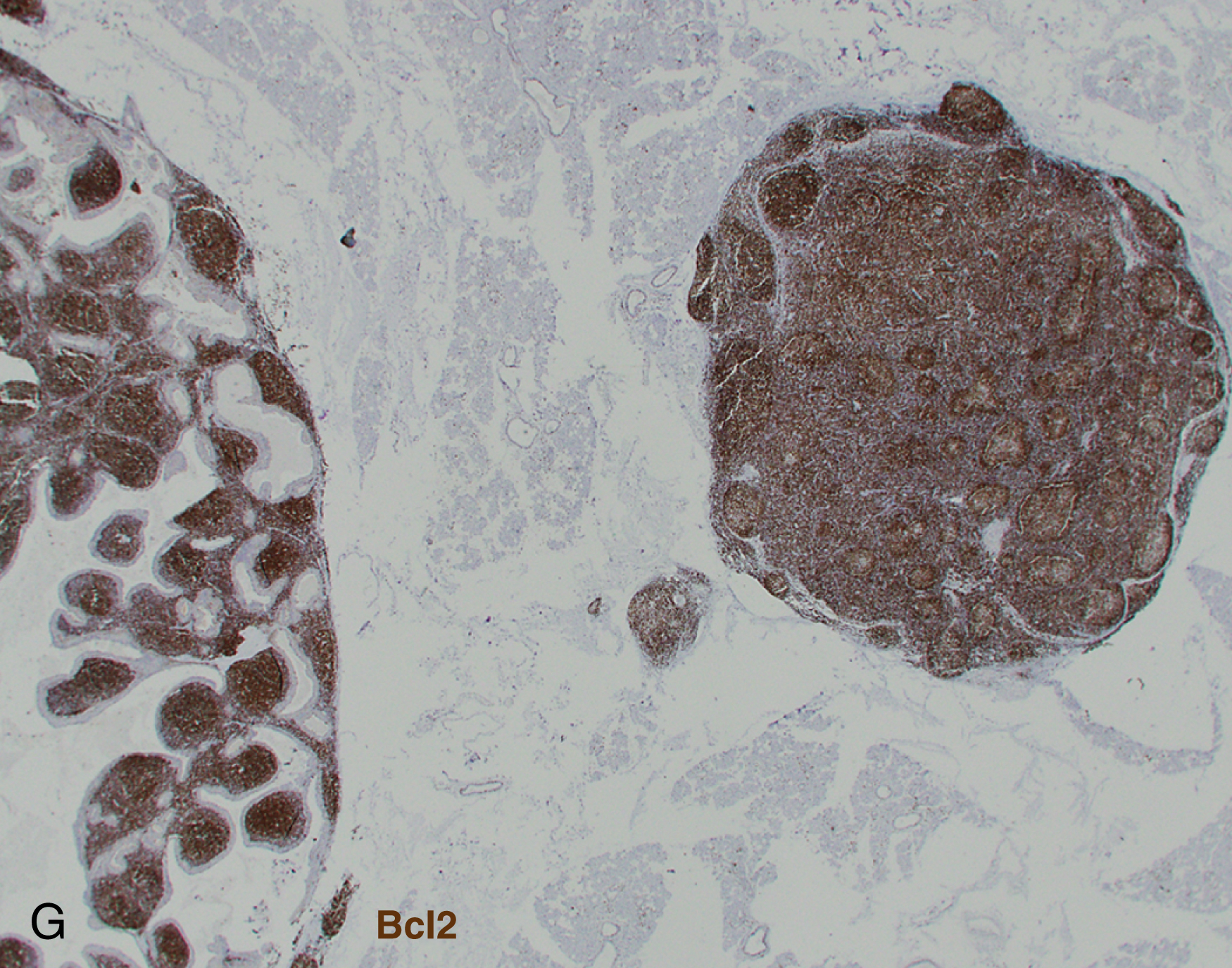
Pediatric-type follicular lymphoma is now a definite entity in the revised 2016 WHO monograph. , , Histologic features are characterized by large, expansile proliferative follicles composed of cells that may be blastoid as opposed to centroblasts or centrocytes with a high proliferation rate ( Fig. 12.3 ). The disease presents most often in lymph nodes, less frequently in the Waldeyer ring or testes, and generally with low clinical stage. The IGH/BCL2 translocation is absent as are BCL6 or MYC translocations, but these cases are generally positive for IGH/IGK clonal rearrangement. Cases involving the Waldeyer ring may be distinguished by the lack of MUM1 expression. Strong positivity for IRF4/MUM1 should raise the possibility of a diagnosis of large cell lymphoma with IRF4 rearrangement.
Primary Cutaneous Follicle Center Lymphoma
Primary cutaneous follicle center lymphoma is the most common type of primary cutaneous B-cell lymphoma. It often presents on the scalp, forehead, or trunk and has a median age at presentation of 51 years with a slight male predominance. In contrast to follicular lymphoma, it frequently lacks the BCL2 translocation as well as BCL2 expression. Indeed, when BCL2 expression is detected, the possibility that this may represent a secondary site of involvement should be considered. These lesions generally have a low risk of spread beyond the skin.
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
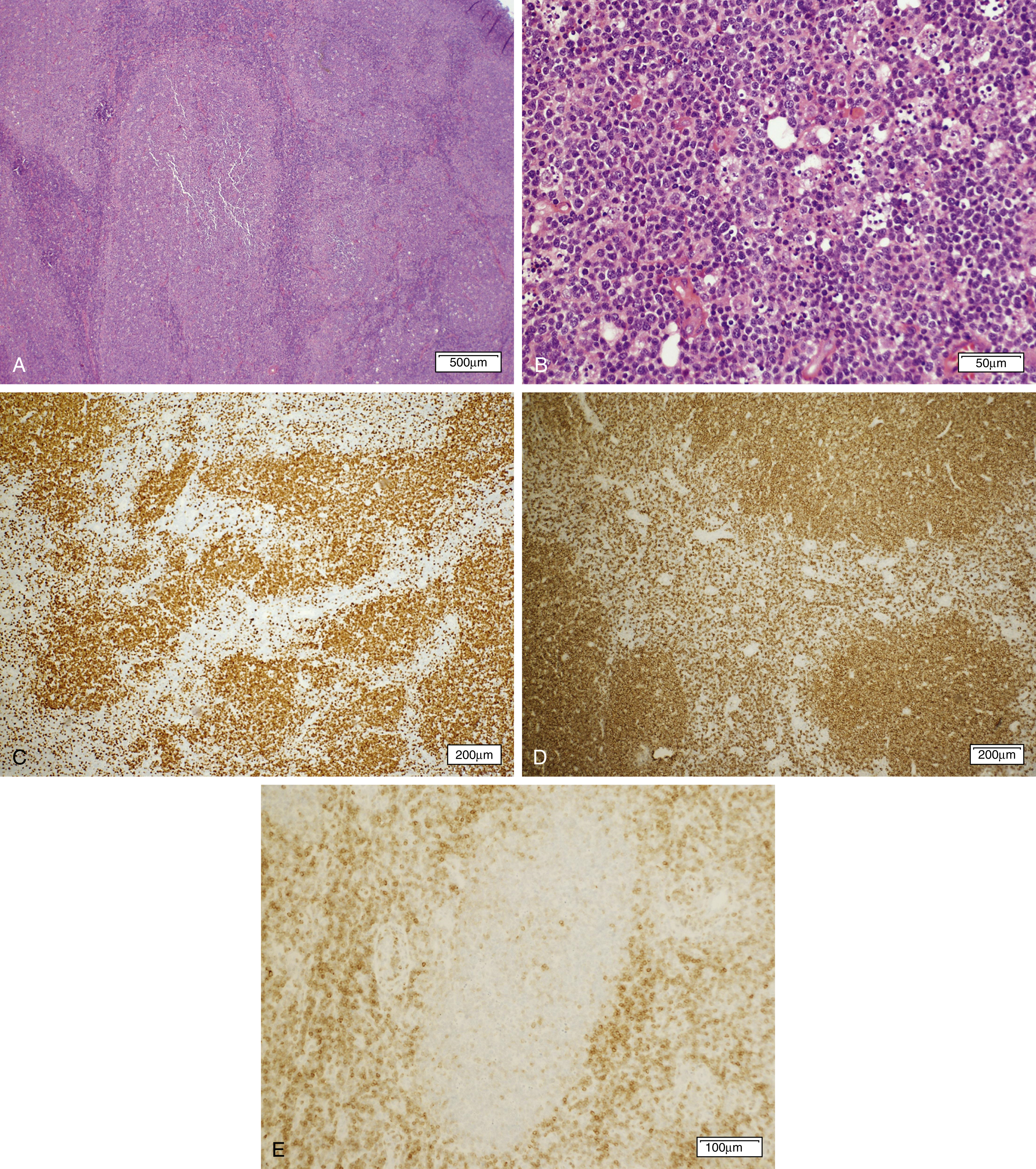
Chronic lymphocytic leukemia and small lymphocytic lymphoma (CLL/SLL) ( Fig. 12.4 ) are the same disease and are primarily considered in Chapter 10 . Nonetheless, a brief discussion of some of the key aspects of the lymphomatous form of the disease is covered herein.
Significantly involved lymph nodes show architectural effacement by small lymphocytes with scant cytoplasm and clumped chromatin. The infiltrate typically has a vaguely nodular appearance with some lighter areas corresponding to proliferation centers, which may show dim cyclin D1 expression and represent a continuum from small lymphocytes to prolymphocytes (increased cytoplasm and prominent nucleoli) to paraimmunoblasts. A tissue-based form of monoclonal B-cell lymphocytosis is also now being recognized when lymphadenopathy is less than 1.5 cm and involved nodes lack evidence of proliferation centers. ,
Alternatively, expansion of proliferation centers (reaching confluency or size greater than a ×20 field) is associated with a more aggressive disease course. Frank transformation to diffuse large B-cell lymphoma, so-called Richter transformation , may occur in 2% to 8% of cases, with more rare transformation to classic Hodgkin lymphoma (<1%). The transformed component may be clonally related to the underlying CLL clone (78% of DLBCL in one large series ) or a separate clonal proliferation. Epstein-Barr virus (EBV)-associated lymphoproliferative processes, including EBV-associated classic Hodgkin lymphoma, may also arise in the setting of CLL/SLL treatment with immunosuppressive agents including fludarabine. , Scattered Hodgkin–Reed-Sternberg (RS) cells may also arise in CLL/SLL ( Fig. 12.5 ). Therefore, diagnosis of transformation requires the presence of RS cells in the appropriate background ( Fig. 12.6 ).

Mantle Cell Lymphoma
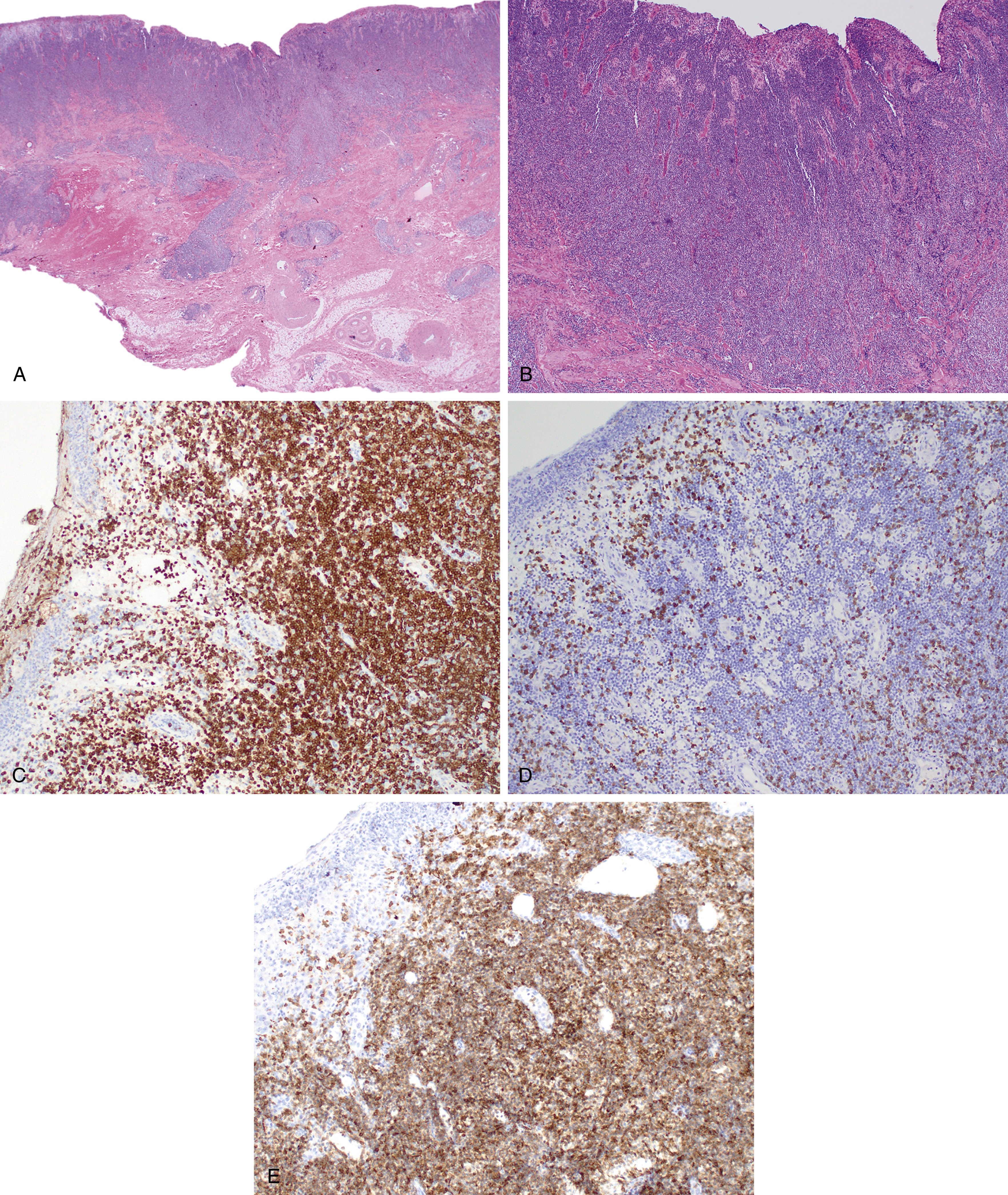
Mantle cell lymphoma (MCL) comprises 3% to 10% of NHLs and usually presents in middle aged to older adults with a median age at presentation of 63 years and male predominance , ( Fig. 12.7 ). MCL is composed of small lymphoid cells with slightly irregular nuclear contours, finely clumped chromatin, and scant cytoplasm. Blastoid and pleomorphic variants have been associated with a more aggressive clinical course ( Fig. 12.8 ). Although mantle cell lymphoma is characterized by the presence of a t(11;14)(q13;q32) translocation, numerous other genetic mutations occur, and there are additional nonrandom secondary chromosomal abnormalities, including loss or mutation of TP53 . , In MCLs that are negative for the t(11;14) translocation and cyclin D1 expression, SOX11 positivity may aid in diagnosis. Otherwise, the genetic landscape and additional mutations associated with these disorders appear to be similar. The proliferation rate based on Ki67 positivity in MCL has been considered to be of prognostic relevance.
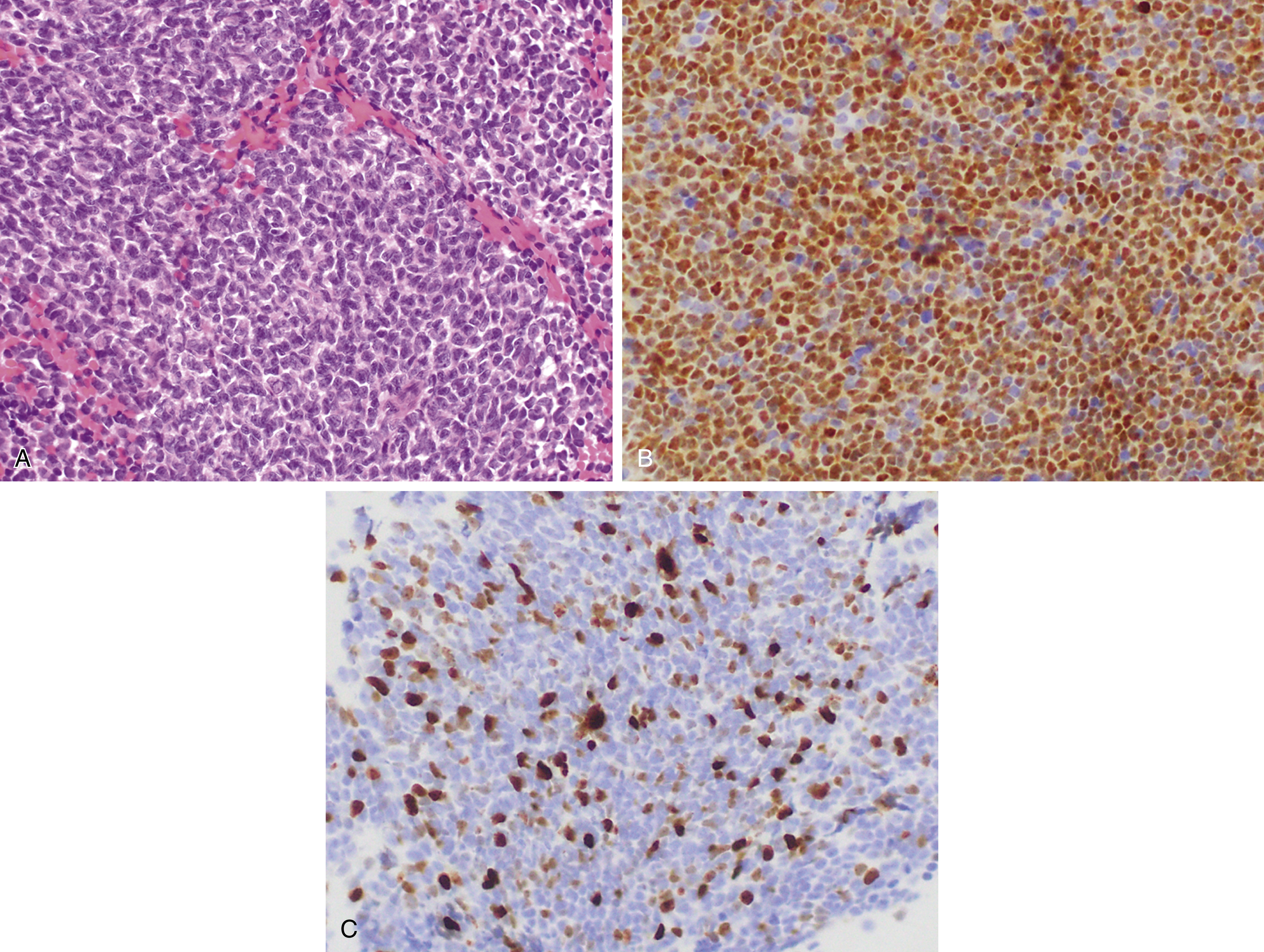
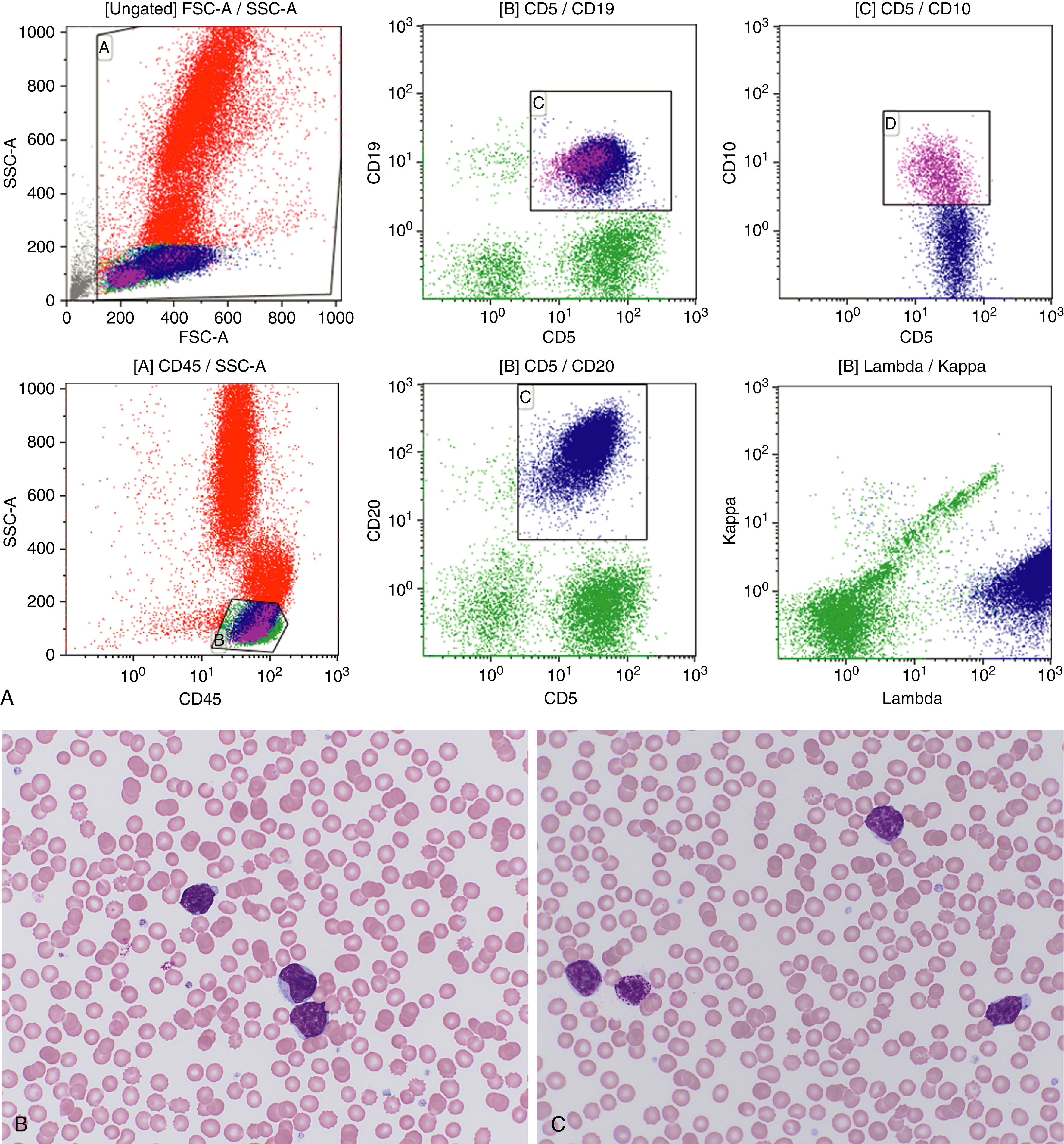
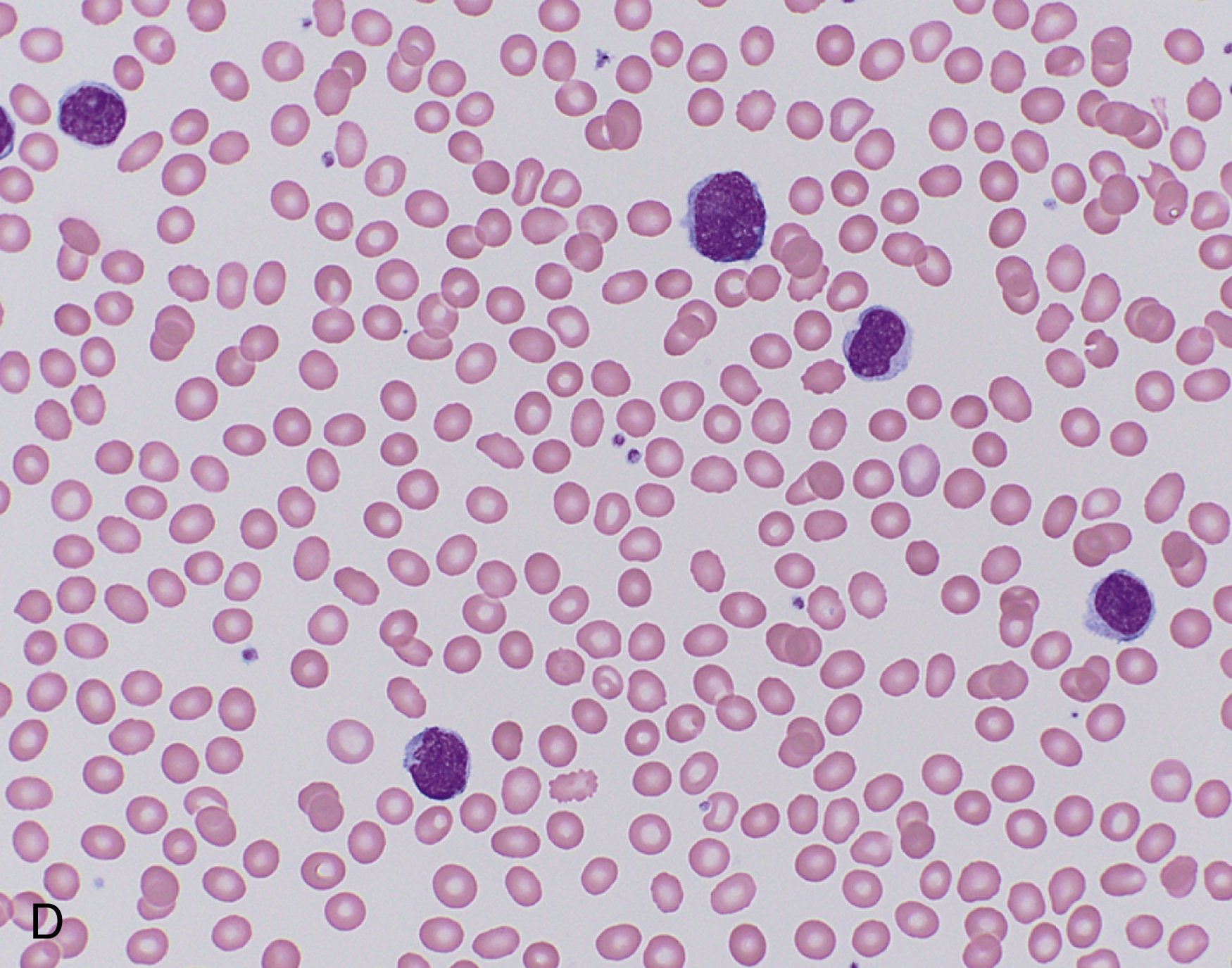
In situ mantle cell neoplasia (ISMCN, according to the new 2016 classification ; see Table 12.2 ) is used to describe a clonal proliferation of cyclin D1–positive cells restricted to the inner mantle cuffs in an otherwise reactive lymph node/lymphoid tissue, and usually represents an incidental finding. Some cases eventually progress to overt MCL. A second indolent variant is characterized by a leukemic phase without nodal disease but often long-standing splenomegaly. Although they carry t(11;14) translocations, they have few additional chromosomal abnormalities. The lack of SOX11 expression combined with the clinical presentation can aid in diagnosis.
Marginal Zone Lymphoma
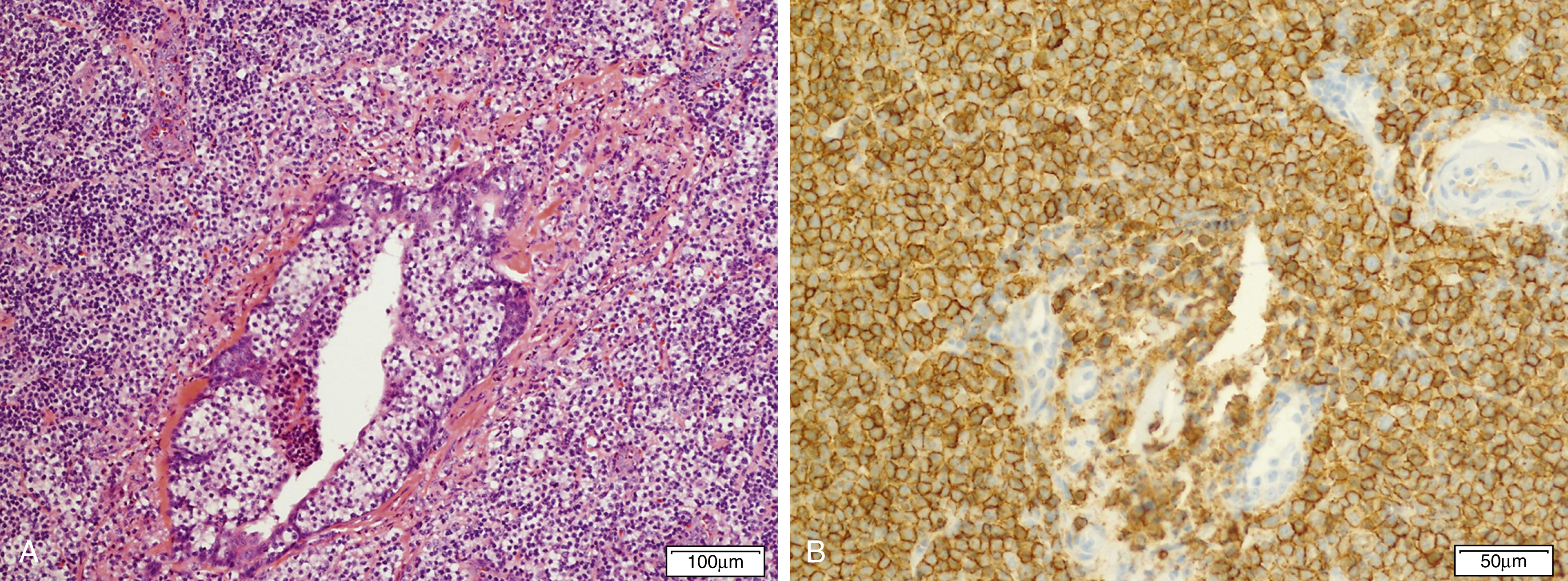
Marginal zone lymphomas include extranodal marginal zone lymphomas of mucosa-associated lymphoid tissue (MALT lymphoma) ( Fig. 12.9 ), , , nodal marginal zone lymphoma ( Fig. 12.10 ), and splenic marginal zone lymphoma. , Marginal zone lymphoma is discussed in more detail in Chapter 10 . Key molecular and cytogenetic data for MZL are highlighted in Table 12.3 .
Small B-Cell Lymphomas With Splenic Involvement
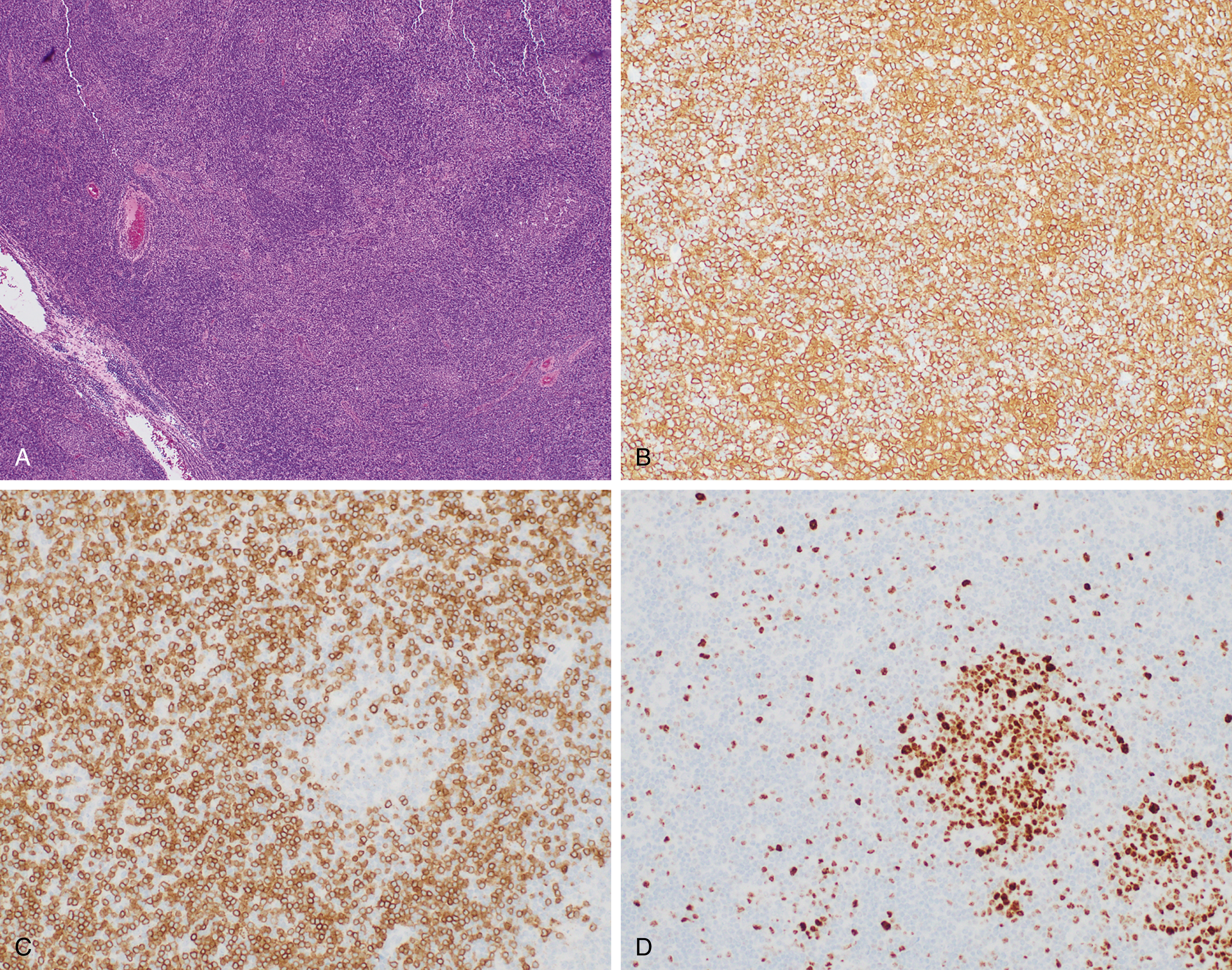
The diagnostic criteria for small B-cell lymphomas with splenic involvement are evolving. These include hairy cell leukemia and variant hairy cell leukemia, which are covered in the chapter on chronic leukemia (see Chapter 10 ) as well as splenic marginal zone lymphoma. Splenic diffuse red pulp small B-cell lymphoma has only recently been identified as a distinctive entity and remains a provisional entity in the 2016 revised WHO monograph. The median age at presentation is 65 years with a male predominance. Patients characteristically have a leukemic presentation with villous lymphocytes and massive splenomegaly. The spleen shows diffuse involvement by small B cells, which involve the cords and sinuses but spare follicular structures. The neoplastic cells are typically CD20 + positive with frequent expression of DBA44 + and IgG, but negative for CD23, BCL6, and annexin A1. Because it shares some overlapping features with splenic marginal zone lymphoma, a diagnosis of splenic diffuse red pulp small B-cell lymphoma may require examination of the spleen ( Fig. 12.11 ). Loss of TP53 or 7q deletion have been reported in a subset of cases (2/13 and 1/13, respectively ). The BRAF V600E mutation characteristic of hairy cell leukemia is not identified.

Lymphoplasmacytic Lymphoma
Lymphoplasmacytic lymphoma (LPL) is a disease of adult life (median age 63–68 years) and usually presents with generalized lymphadenopathy, constitutional symptoms, and splenomegaly with a slight male predominance. In particular, patients tend to present with fatigue related to anemia. The postulated normal counterpart is thought to be a post-follicular B-cell type based in part on the presence of somatic mutations in the immunoglobulin (Ig) Ig heavy- and light-chain–variable region genes.
Histologically there is a diffuse interfollicular proliferation of small lymphocytes, including many with plasmacytoid features and plasma cells with general sparing of the sinuses. Increased numbers of mast cells and iron-laden macrophages can be seen. Although many B-cell neoplasms occasionally show maturation to plasmacytoid/plasma cells, the term LPL should be restricted to tumors lacking features of other well-defined entities (e.g., chronic lymphocytic leukemia or MCL) that occasionally can manifest plasmacytoid differentiation. Many patients with LPL may have clinical evidence of Waldenström macroglobulinemia (WM), which is based on the detection of an IgM monoclonal gammopathy of any concentration associated with bone marrow involvement by LPL. LPL is generally negative for CD5 and cyclin D1, but CD25, CD10, or CD11c may be weakly expressed in some cases.
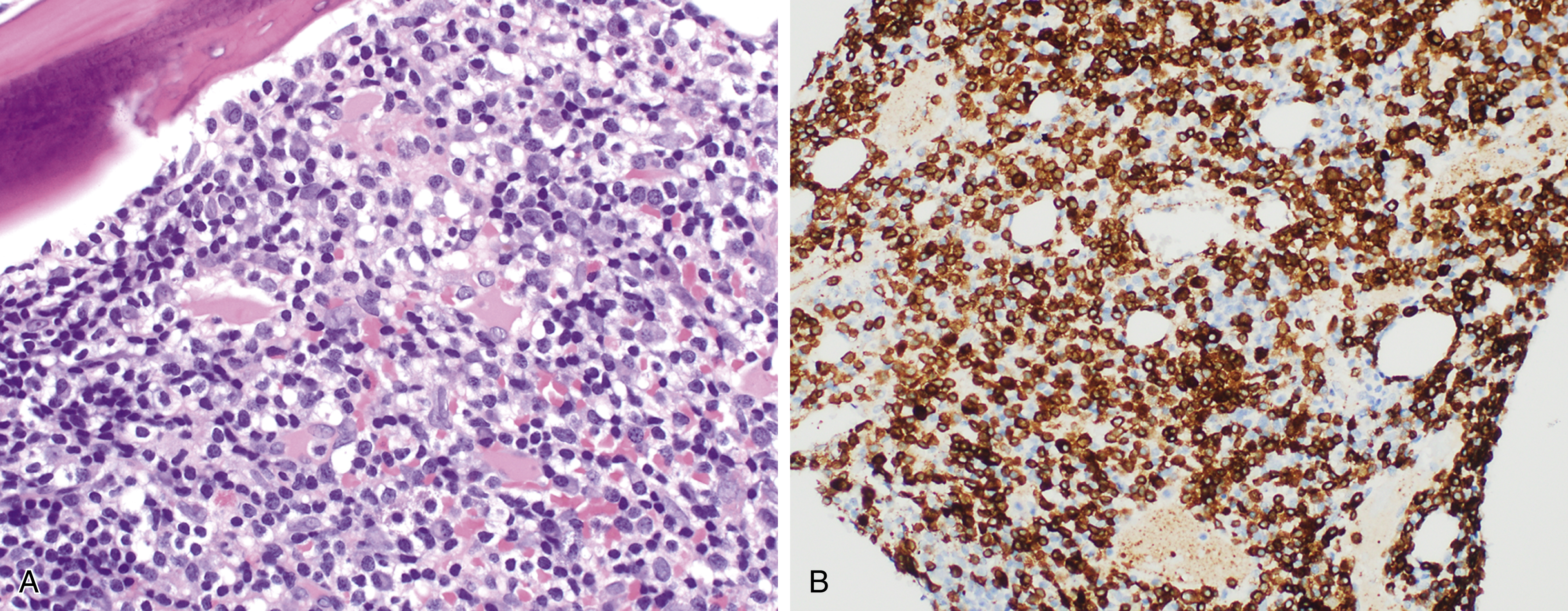
More than 90% of LPL and WM cases have an MYD88 L265P mutation, which may also be present in IgM monoclonal gammopathy of uncertain significance (MGUS) and certain subtypes of diffuse large B-cell lymphoma (see later text). , , Mutations in the cysteine-X-cysteine chemokine receptor 4 (CXCR4) are found in 30% of cases of LPL and 20% of cases of IgM MGUS. CXCR4 mutations are also associated with the syndrome of warts, hypogammaglobulinemia, infections, myelokathexis (WHIM syndrome), caused by impaired ability of neutrophils and other white blood cells to be able to egress from the bone marrow. , Similarly, patients with CXCR4 mutations in the context of LPL showed a higher extent of bone marrow infiltration and lower leukocyte counts ( Fig. 12.12 ).
Aggressive B-Cell Neoplasms
Burkitt Lymphoma
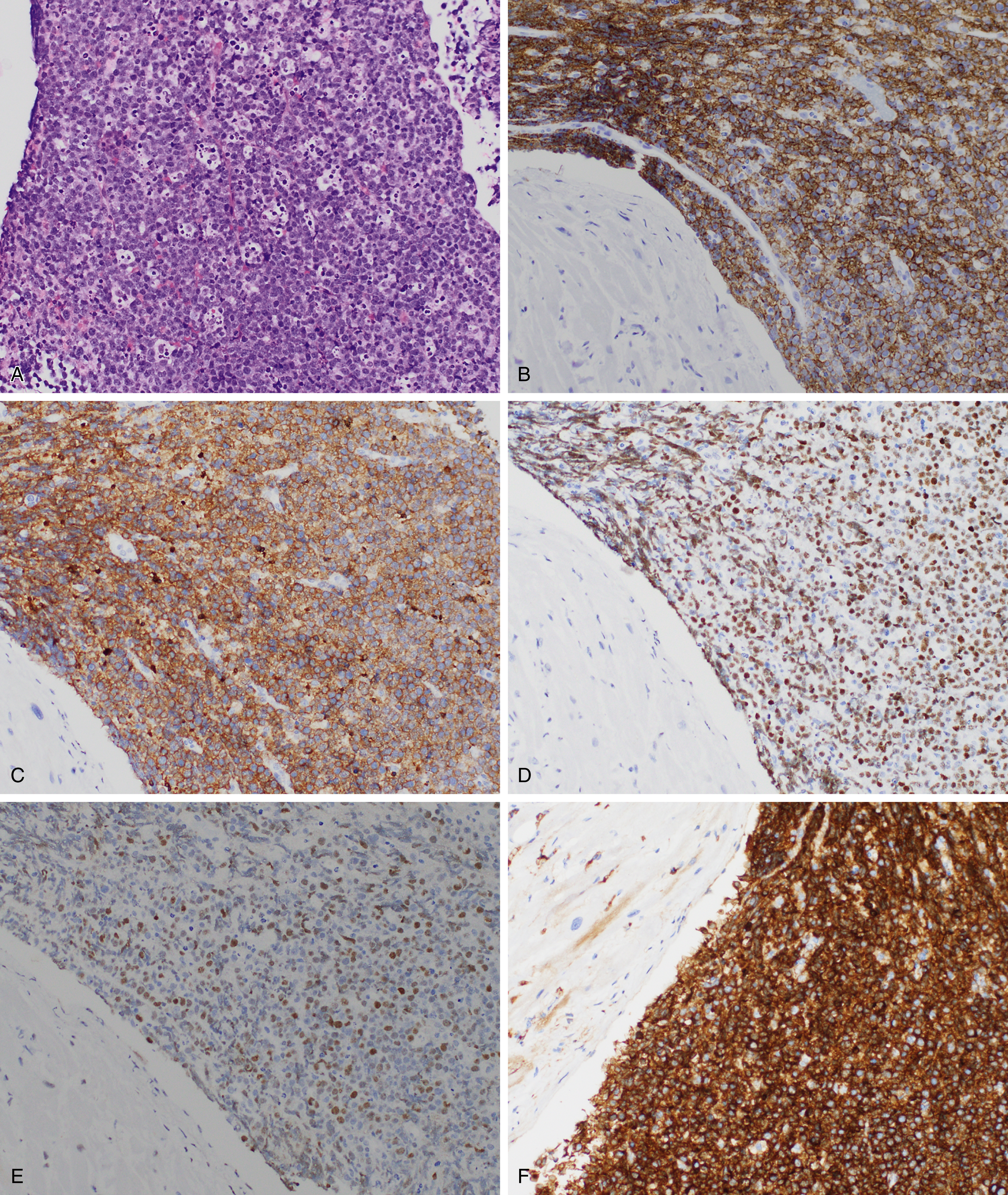
Burkitt lymphoma is most common in children and accounts for up to one-third of all pediatric lymphomas in the United States. Three clinical variants of Burkitt lymphoma are recognized: endemic, sporadic, and immunodeficiency associated. The endemic cases are seen in equatorial Africa in regions endemic for malaria and mostly involve the jaw and other facial bones. In nonendemic regions, such as the United States, extranodal sites are more frequently involved, including the ileocecal region, ovaries, kidneys, or breasts. A patient with disease-isolated cardiac involvement is shown in Fig. 12.13 . Burkitt lymphoma is one of the more common tumors associated with the human immunodeficiency virus (HIV). It can be the initial AIDS-defining illness but can present at any time during the clinical course. Bone marrow involvement is a poor prognostic sign. EBV has been shown as a cofactor for the development of Burkitt lymphoma and shows varying degrees of positivity in the clinical variants.
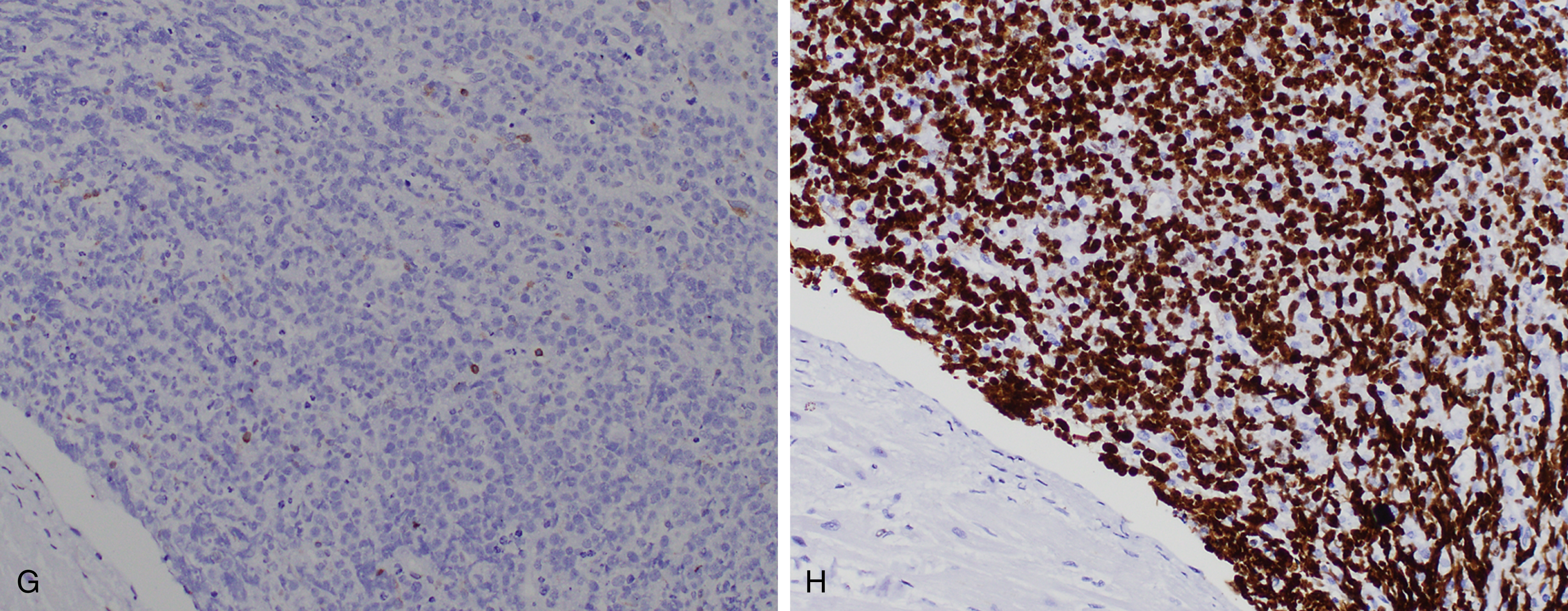
Burkitt lymphoma is defined in part by the presence of an MYC translocation, which juxtaposes MYC with one of the three immunoglobulin loci (t(8;14)(q24;q32), t(2;8)(p12;q24), or t(8;22)(q24;22q11)), but these also may occur in DLBCL (10% of cases) , ( Table 12.4 ). In addition, MYC perturbation alone does not appear to be sufficient for Burkitt lymphomagenesis, although the overall karyotype of Burkitt lymphoma tends to be simple. Of interest, mutations in ID3 , a negative regulator of TCF3 (which is overexpressed in Burkitt lymphoma and a transcriptional target of MYC ), appears to be an additional and relatively specific driver mutation. , Other genes involved include ARID1 and SMARCA4 (SWI/SNF pathway) and GNA13 and RHOA (focal adhesion pathway). Burkitt lymphoma, as defined by a molecular signature, has also been described with gains of chromosome 1q being common.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


