Chapter 17 Indinavir
Indinavir sulfate (IDV, L-735,524; MK-639; Crixivan) is a human immunodeficiency virus type 1 (HIV-1) protease inhibitor (PI). The drug was approved by the US Food and Drug Administration (FDA) in March 1996 and is labeled for the treatment of HIV infection in combination with other antiretroviral agents.1 Current antiretroviral guidelines no longer recommend IDV (with or without ritonavir (RTV) boosting) as initial therapy for HIV disease.2,3 IDV/RTV may be used in some treatment-experienced patients.4,5
STRUCTURE
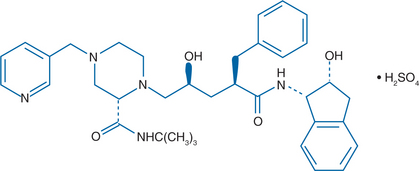
The chemical name for IDV sulfate is [1 (1S, 2R),5 (S)]-2,3,4-trideoxy-N-(2,3-dihydro-2-hydroxy-1H-inden-1-yl)-5-[2-[{(1,1-dimethylethyl) amino}carbonyl]4-(3-pyridinylmethyl)-1-piperazinyl]-2-(phenylmethyl) D-erythro-pentonamide sulfate (1:1) salt (Fig. 17-1).1,6 IDV is derived from the class of hydroxyethylene peptidomimetic PIs that demonstrate potent antiretroviral activity but have poor aqueous solubility in vitro and inadequate oral bioavailability in animal models.6 By incorporating a basic amine group into the structural backbone of the hydroxyethylene PIs, a new class of PIs, the hydroxyethylamine compounds, were developed. This class of compounds retains its peptide character but has improved aqueous solubility and enhanced oral bioavailability. Further improvements were achieved using a rational structure-based drug design to model and develop a series of structural analogs of these compounds in an attempt to retain potent antiretroviral activity while further improving oral bioavailability.6–8 The modified hydroxyethylamine peptidomimetic compounds are exemplified by the HIV PI IDV.
IN VITRO ACTIVITY
IDV potently and competitively inhibits both the HIV-1 and HIV-2 aspartyl protease enzymes with KI (dissociation constant of the enzyme-inhibitor complex) values of 0.34 and 3.3 nM, respectively.9 In concentrations higher than 10 μM, IDV did not inhibit other clinically relevant proteolytic enzymes such as the aspartyl proteases human plasma renin or human cathepsin D or the serine proteases factor Xa or elastase. IDV is a potent inhibitor of HIV-1 replication in vitro.9 In cell culture systems, IDV showed potent activity against T lymphoid cell-adapted HIV variants (IIIb, MN, RFII) and a monocytotropic variant (SF162) with a 95% inhibitory concentration (IC95) in the range of 12–100 nM. IDV also demonstrated potent activity against primary patient HIV isolates from peripheral blood mononuclear cells, including isolates with zidovudine or non-nucleoside reverse transcriptase inhibitors (NNRTI) resistance. IDV showed synergistic in vitro antiviral activity in combination with zidovudine, didanosine, or an NNRTI. The combination of IDV and other PIs showed synergism to mild antagonism in vitro.10 IDV demonstrated activity in vitro against HIV-211,12 and the simian immunodeficiency virus.11
MECHANISM OF ACTION
The HIV protease enzyme cleaves viral precursor polyproteins into structural proteins and enzymes, a step essential for the production of mature, infectious virions.13 Retroviral proteases are unusual among proteolytic enzymes in terms of their ability to cleave protein substrates at the N-terminal side of proline residues. Phenylalanine–proline (Phe-Pro) residues constitute one of the important cleavage sites for the HIV-1 protease enzyme. The hydroxyethylamine peptidomimetic PIs, including IDV, were designed to mimic the Phe-Pro dipeptide transition state of the substrate protein.6–8 These inhibitors enter the cell without intracellular processing, bind at the active site of the protease enzyme, inhibit cleavage of the viral precursor proteins, and thereby prevent maturation of the virus. Immature, noninfectious viral particles are formed in the presence of the peptidomimetic PIs. The crystal structure of IDV complexed to the HIV protease shows that binding causes closure of the flap domains of the enzyme. There are specific interactions between chemical groups of the inhibitor and the enzyme, with the lipophilic groups of IDV (except the pyridine ring) confined inside the active site of the enzyme.14
Adding IDV at concentrations of 500–12 000 nM to chronically HIV-infected cell culture systems decreased the amount of mature HIV proteins (p17 and p24) and increased the amount of the viral core protein precursor (p55) compared to untreated virions.9 IDV also prevented incorporation of the HIV reverse transcriptase and integrase enzymes into viral particles. In parallel experiments, removing IDV from the cell culture medium resulted in an increase in the amount of mature viral proteins and the production of infectious virions. Thus, IDV is a reversible inhibitor of the HIV protease enzyme.
In animal studies, IDV showed antiangiogenic effects and promoted regression of Kaposi sarcoma-like lesions.15
PHARMACOKINETICS
Preclinical Profile
In animal models the plasma concentrations 6 h after administration of oral IDV solutions were twice the in vitro IC95.6,9 Oral bioavailability ranged from 14% in monkeys to 72% in dogs. IDV required an acidic pH environment for dissolution prior to absorption from the gastrointestinal tract and was less soluble at higher pH.16 Other in vitro studies showed that IDV was not highly bound to plasma proteins, with the unbound drug fraction ranging from 15% in dog plasma to 56% in human plasma at drug concentrations of 81–16 300 nM.6,9 In contrast to other PIs, the antiretroviral activity of IDV in vitro was similar against wild-type and PI-resistant isolates despite a fourfold increase in α1-acid glycoprotein levels.17 By inhibiting hepatic metabolism and clearance, RTV enhances the levels of other PIs, including IDV.18 With concomitant RTV administration, IDV area under the curve concentration (AUC) was increased eightfold in rats.
CLINICAL PROFILE OF IDV
The first clinical study of IDV was an assessment of the safety, tolerability, and pharmacokinetics of single doses in 28 healthy volunteers.19 Subjects were administered doses ranging from 20 to 1000 mg of the free base formulation of IDV. Plasma concentrations and urinary excretion were nonlinear, increasing more than proportionally to dose. The plasma half-life of the drug averaged 1.8–1.9 h and was not dose-dependent. Calculated free drug concentrations exceeded the in vitro IC95 through 6 h after the 700-mg dose and through 8 h following the 1000-mg dose.
To investigate the nonlinear pharmacokinetics further, 12 healthy volunteers were administered single intravenous and oral doses of IDV.20 Although at low plasma concentrations IDV clearance was high, hepatic first-pass metabolism appeared to be saturable at higher doses of the drug (400- and 800-mg oral doses), resulting in reduced drug clearance and higher drug concentrations. In this study, oral bioavailability was estimated to be 60–65%. An intensive pharmacokinetic study in eight HIV-infected adults showed overall IDV protein binding of 61 ± 6% (range 54–70%), with considerable variability among patients.21 The fraction of bound drug was concentration-dependent in that IDV binding was higher at the 8-h postdose concentration than at the 1-h time point.
In dose-escalation studies, IDV was given at doses of 800 mg q8h, 1000 mg q8h, and 800 mg q6h in a study of 70 HIV-positive adults.22 The geometric mean plasma AUCs over 24 h for the three regimens were not statistically different and the 800-mg q8h dose was selected as the optimal dose. In 16 patients at the 800-mg q8h dose, the steady-state AUC was 30 691 ± 11 407 nM/h, the Cmax was 12617 ± 4037 nM, and the Cmin 8-h postdose was 251 ± 178 nM.1 There was no significant accumulation with multiple doses at 800 mg q8h. Alternative dosing of IDV at 1200 mg q12h was compared to 800 mg q8h in a combination regimen and found to have inferior virologic activity (64% vs 91% of subjects respectively had an HIV RNA level of less than 400 copies/mL at week 24) likely due to suboptimal pharmacokinetics, and this dose schedule is not recommended.23
In 17 patients taking IDV- or IDV/RTV-based regimens, median IDV protein binding was 64% without differences with or without RTV.24 Another group studied 35 patients taking IDV/ritonavir at doses of 800/200 or 400/400 mg bid and found IDV was ∼52% protein-bound without differences between the groups, but with significantly higher binding at peak concentrations, suggesting concentration-dependence of protein binding.25
FOOD EFFECTS
Administration with food high in calories, fat, and protein reduced the absorption of IDV.19 In eight HIV-infected subjects, a 600-mg dose of IDV given with food significantly decreased absorption 30–68% compared with fasted controls with reductions in Cmax and a delayed time to Cmax.26 In clinical studies of HIV-uninfected volunteers, various dose combinations of IDV/RTV were studied and adding RTV allowed twice-daily IDV dosing with food.27,28 To explore the effects of food on IDV/RTV, nine HIV-infected patients taking IDV 800 mg/RTV 100 mg bid for at least 4 weeks were randomized to take the combination with a light meal or fasting and underwent pharmacokinetic evaluation.29 Taking IDV/RTV in a fasted stated resulted in a 1.3-fold higher IDV Cmax compared to administration with food (p = 0.001), without changes in AUC or Cmin. The investigators hypothesized that food induced a delay in IDV absorption and suggested this as a strategy to avoid the high Cmax concentrations that have been linked to IDV-related nephrotoxicity.
CLINICAL PROFILE OF IDV/RTV
Based on pharmacokinetics and tolerability, the optimal IDV/RTV dose appears to be 800/100 mg twice daily; this strategy increased the IDV AUC 2.3-fold and the Cmin 10–25-fold.28 In two clinical studies in HIV-infected subjects, the combination of IDV 800 mg/RTV 100 mg twice daily also was found to have favorable pharmacokinetic properties.30,31 Other doses also have been explored. Solas et al reported 17 HIV-infected patients taking IDV 400 mg/RTV 200 mg bid increased the IDV Cmin and lowered Cmax, but was associated with toxicity.32 Boyd and colleagues reported 19 Thai patients taking IDV 400 mg/RTV 100 mg bid but found significantly reduced AUC by 63%, Cmax by 61% and Cmin by 76%.33 Wasmuth studied 16 HIV-negative volunteers who took IDV 600 mg or 400 mg/RTV 100 mg bid and reported worse tolerability in the 600-mg group, but significantly lower IDV exposure in the 400-mg group.34 Cressey et al studied 13 HIV-infected, treatment-naive patients who started an IDV 600 mg/RTV 100 mg bid-based regimen and then reduced to IDV 400 mg after 1 month.35 The 600 mg dose was associated with toxicity, while the 400 mg dose was associated with reductions in AUC of 54%, Cmax by 39%, and Cmin by 59%. Rhame compared 27 HIV-infected patients randomized to receive IDV 800 mg q8h (standard dose) versus IDV 667 mg/RTV 100 mg bid and found the RTV-boosted dosing was associated with an increased IDV AUC by 1.5-fold, Cmin by sixfold with no change in Cmax.36 Once-daily dosing of IDV/RTV also has been explored but is limited by tolerability.37–40 Zala et al described 10 patients taking an IDV 800 mg/RTV 100 mg bid-based regimen who substituted a generic formulation of IDV made in Argentina (Inhibisam, Laboratrio Richmond, Buenos Aires) and found no significant differences in IDV exposure.41
ELIMINATION
In one of the initial clinical studies, 1000-mg oral doses of IDV were administered to 10 healthy volunteers, and urine samples were collected and analyzed.19,42 Seven major metabolites were identified, and glucuronidation, oxidation, and N-alkylation were the major metabolic pathways. The major component of drug in urine was unchanged IDV, accounting for 11% of the total dose. In follow-up experiments, single 400-mg oral doses of radiolabeled IDV were given to six healthy volunteers.43 The major route of excretion of IDV was via feces (19% unchanged drug, 64% metabolites); the minor excretory route was via urine (9% unchanged drug, 10% metabolites); and the combined recovery was quantitative. Seven drug metabolites were identified in both plasma and urine; six oxidative metabolites and one glucuronide conjugate. The high level of metabolites in feces is consistent with biliary excretion. The cytochrome P450 (CYP) 3A4 hepatic isoform is the major enzyme responsible for formation of the oxidative metabolites44; intestinal CYP3A4 likely contributes relatively little to its metabolism.45 IDV (like other PIs) serves as a substrate for the P-glycoprotein (pgp) efflux membrane transporter, which is distributed in the gastrointestinal tract, liver, kidney, blood–brain barrier, genital tract, and some CD4+ T lymphocytes.46 The cellular pump may promote drug secretion into the intestinal lumen, enhance metabolism, or block entry into brain, testes, and some CD4+ T lymphocytes.
TISSUE PENETRATION AND RESERVOIRS
After oral administration in rats, IDV was distributed rapidly throughout the plasma and lymphatic system, with comparable concentrations.47 Intracellular lymphocyte concentrations in HIV-infected patients are not significantly different from plasma concentrations although the intracellular elimination half-life and mean residence time of IDV were prolonged.48 Solas et al studied 41 patients on stable IDV-based regimens and found a twofold higher IDV concentration in lymphoid tissue compared to plasma.49
In vitro and animal experiments showed that IDV was transported by a pgp efflux membrane transporter that is thought to limit brain (and other tissue) penetration.46 Following intravenous administration to rats, the brain-plasma IDV-level ratio was 0.18 at steady state, suggesting limited penetration.47 In an intensive clinical study, eight HIV-infected adults who took an IDV-containing regimen underwent sampling of cerebrospinal fluid (CSF) and plasma.50 The mean values of free IDV in the CSF were AUC (0–8 h) 1616 nm/hr, Cmax 294 nM, and Cmin 122 nM, all exceeding the in vitro IC95 for IDV. Free IDV accounted for 94% of the drug in CSF and 42% in plasma. The mean ± SD CSF/plasma ratio for free IDV was 15 ± 3%. Seven of eight subjects had CSF HIV RNA levels of less than 200 copies/mL. Other population-based pharmacokinetic studies have shown similar results.49,51–53
CSF levels of IDV were higher when administered with RTV.54,55 For example, in a pilot study of 13 HIV-infected patients, the addition of RTV to IDV increased IDV concentrations in the serum (Cmin from 65 to 336 ng/mL), CSF (from 39 to 104 ng/mL), and seminal plasma (median 141–1634 ng/mL).54 In six patients with before and after samples, RTV increased IDV levels significantly in CSF 2.4-fold and in seminal plasma by 8.0-fold. Another group found similar findings in CSF in seven patients taking IDV/RTV.55
Solas and colleagues found IDV concentrations were 1.9-fold higher in semen than plasma.49 Also, Taylor et al found median seminal to plasma IDV concentration ratios ranged from 0.6 (0–2-h postdosing) to 1.4 (6–8-h postdosing), although the concentrations exceeded the IC95 at all time points.56 Van Praag et al characterized IDV concentrations in two fractions of seminal plasma (testicular-prostate, seminal vesicle) and concluded IDV achieved therapeutic concentrations in both throughout the dosing interval.57 Several groups have documented HIV RNA reduction in semen in patients taking IDV-containing regimens.58,59
Launay et al studied nine women taking IDV-based regimens and found IDV detectable in 93% of cervicovaginal secretions.60 In an ex vivo assessment of placental transport, maternal-to-fetal clearance of IDV was significantly lower than fetal-to-maternal clearance.61 In a study of a nursing mother taking an IDV-based regimen, breast milk drug concentrations were 90–540% of her plasma concentrations.62
PHARMACOKINETICS AND ANTIRETROVIRAL EFFECT
Several groups have related the plasma concentrations of IDV to its virologic effect.63–68 Burger and colleagues prospectively followed 65 subjects on an IDV-containing regimen and observed a virologic failure rate of 37%.63 In a multivariate analysis, a low plasma concentration of IDV was an independent predictor of failure. Similarly, Acosta and colleagues studied 43 subjects receiving IDV combination therapy and found significantly higher IDV AUC (0–8 h) in treatment-naive subjects with HIV RNA below the limit of detection, compared to those with detectable viral load levels.64 In another study, using modeling in a treatment-naive patient population, higher IDV Cmin values were significantly associated with virologic suppression to less than 200 copies/mL at week 24.65 The 90% virologic response rate in this study correlated with an IDV Cmin of 110 ng/mL. Anderson and colleagues correlated higher IDV Cmax concentrations (>7 μg/mL) with greater CD4+ T lymphocyte increases in a group of patients with suppressed HIV RNA levels.66 Duval et al studied 216 patients taking IDV-based regimens and found that virologic response was independently associated with full adherence, but not IDV concentration in a multivariate model (OR 8.8, P < 0.05).67 A Spanish group studied 46 patients experiencing early virologic failure on an initial IDV-based regimen and found 69% had subtherapeutic IDV levels (75% of whom had no detectable levels) and found more IDV-related resistance mutations in the group with suboptimal, but detectable IDV concentrations.68
Csajka and colleagues conducted a population pharmacokinetic analysis of 239 patients taking IDV with or without RTV and demonstrated significant interpatient and intrapatient variability in IDV concentrations and suggested this supported therapeutic drug monitoring (TDM).69 Kakuda et al showed the safety and feasibility of concentration-controlled therapy in a pilot study of 11 subjects receiving an IDV-based combination regimen with dosing targeted to achieve a target trough IDV level of 0.15 mg/L.70
Shulman and colleagues reported 37 patients taking an IDV-based regimen with ongoing viremia who were changed to a regimen of IDV 400 mg/RTV 400 mg bid and underwent pharmacokinetic assessment.71 The IDV/RTV regimen did not change IDV AUC, but decreased Cmax by 57% and increased the Cmin 3.4-fold and resulted in 21 (58%) of patients reducing HIV RNA levels to <50 copies/mL. Upon further analysis, they found a virtual inhibitory quotient (a ratio of the IDV predose concentration to the degree of virtual phenotypic resistance) was a better predictor of response than either drug resistance or drug concentration alone. The Athena study randomized 55 treatment-naive patients to an IDV-containing regimen (with or without RTV) to receive TDM results with dose adjustments based on achieved IDV concentrations versus the standard-of-care (i.e., no TDM or dose adjustment).72 After 1 year, fewer patients in the TDM group discontinued IDV (25% vs 48%, p = 0.07) and there was a better virologic response.
HEPATIC OR RENAL INSUFFICIENCY
Twelve patients with clinical evidence of cirrhosis and mild to moderate hepatic insufficiency who were administered single doses of 400 mg IDV had a 60% higher mean AUC and an increased half-life of 2.8 ± 0.5 h compared to historical controls, indicating decreased metabolism of IDV.1 Based on these results, the recommended dose in patients with mild to moderate hepatic insufficiency resulting from cirrhosis is 600 mg q8h. Bossi and colleagues studied six patients with either chronic hepatitis B or C infection who enrolled in the GENOPHAR study of TDM and were treated with IDV 400 mg/RTV 100 mg bid and reported high plasma IDV concentrations.73 After a dose adjustment at 4 weeks to IDV 200 mg/RTV 100 mg bid, adequate IDV concentrations were maintained through 24 weeks and they suggested this was the optimal dose for co-infected patients. The pharmacokinetics have not been determined in patients with severe hepatic insufficiency or renal insufficiency, but it is assumed that because of the hepatic metabolism of the drug, substantial dose reductions for renal insufficiency are not required.74 It is not known if IDV is dialyzable by peritoneal dialysis or hemodialysis, though case reports suggest that dosage modification may not be necessary.75–77
GENDER AND RACE
Gender differences in IDV metabolism were investigated in the rat, dog, and monkey and in in vitro studies with human microsomes.78 Although some metabolic differences between sexes occurred in the rat and dog, no differences occurred in monkeys or in human liver microsome experiments. Using a population-based analysis of 314 patients, Dutch investigators found no difference in IDV pharmacokinetics by gender although they found dose reductions for IDV-related side effects were more common in women than men (10% vs 1%, P < 0.01) and suggested a subset of 10–20% of women might have higher IDV levels.79 In other clinical studies, comparable pharmacokinetics of IDV were seen in whites and blacks, both in HIV-infected and -uninfected volunteers.1
PEDIATRICS
The optimal dosing of IDV in pediatric patients has not been determined. In a clinical study of 34 pediatric HIV-infected patients (age 4–15 years), IDV dosed at 500 mg/m2 q8h had an AUC (0–8 h) of 38 742 nM/h, Cmax 17 181 nM, and Cmin 134 nM.1 Compared to pharmacokinetic results in adults taking IDV 800 mg q8h, the AUC and Cmax were higher and the Cmin lower. Fifty percent of pediatric patients had levels of less than 100 nM compared to 10% of adults. Uncontrolled studies of IDV 500 mg/m2 q8h in 70 children confirmed different pharmacokinetic profiles from adults.1 Another study of 11 HIV-infected children taking 500 mg/m2 q8h also showed a lower Cmin and suggested that every-6-h dosing is appropriate.80 In addition, these investigators showed that children with a small body surface area had greater AUC values than adults and suggested that dose reduction is appropriate. A Dutch study of 19 children concluded that the optimal dose to obtain IDV exposures similar to adults was 50 mg/kg of metabolic weight q8h (∼600 mg/m2), although they noted some children required higher doses.81 Another Dutch study studied IDV 400 mg/m2/RTV 125 mg/m2 bid in 14 children and reported that the AUC was similar while the Cmax was 20% lower compared to adult dosing of IDV 800 mg/RTV 100 mg bid.82
PREGNANCY
Kosel and colleagues reported the pharmacokinetic evaluation of 4 pregnant women in their second or third trimesters who took IDV 800 mg q8h (n = 2) or IDV 800 mg/RTV 200 mg bid (n = 2).83 In women taking the IDV-based regimens, they found the IDV AUC decreased 52–86% in the third trimester, likely secondary to metabolic induction. However, using IDV/RTV prevented the reduction. Following childbirth, IDV levels returned to baseline.
TOXICITY
In Phase I trials the only clearly drug-related side effect of IDV was a reversible increase in indirect bilirubin.84 Subsequent studies first documented the occurrence of nephrolithiasis.22,85 In addition, studies of combination therapy with IDV and zidovudine first described gastrointestinal side effects.86 In the Swiss HIV Cohort Study of 1160 patients of whom 235 (20%) received IDV, hyperbilirubinemia (odds ratio, OR 18), nephrolithiasis (OR 11) and rash (OR 2), were all significantly associated with IDV use.87 In a French cohort of 1155 patients taking PI-based therapy, 44% of whom took IDV, IDV use was independently predictive of a serious adverse drug reaction (HR 1.7, 95% CI 1.2–2.4) and overall renal colic was the second most commonly reported event (Table 17-1).88
Table 17-1 Common Side Effects of Indinavir
| Side Effect | Incidence (%) |
|---|---|
| Gastrointestinal disturbance | |
| Abdominal pain | 9 |
| Diarrhea | 5 |
| Nausea | 12 |
| Vomiting | 4 |
| Headache | 6 |
| Hyperbilirubinemia (indirect) | |
| Total bilirubin >2.5 mg/dL | 10 |
| Total bilirubin >5.0 mg/dL | 1 |
| Nephrolithiasis (flank pain hematuria) | 9–43 |
OVERDOSE
A 5-year review of the Merck safety data base identified 79 reports of IDV overdose89: 15 single episodes of taking >2400 mg, 13 single extra doses <2400 mg, 43 episodes of taking extra drug doses, and eight with unknown doses. Of these, 52 (66%) had adverse events, most commonly nausea, vomiting, abdominal pain, and nephrolithiasis although most patients recovered.
CRYSTALLURIA AND NEPHROLITHIASIS
In a British cohort study of 781 patients taking IDV-based regimens over a median of 1 year, 7% developed IDV-related renal complications: groin pain, renal colic, and/or dysuria.90 Most patients continued IDV and there was no increase in creatinine. In a Dutch cohort of 1219 patients first starting a PI-based regimen, 644 started IDV and the incidence of urologic symptoms was 8.3/100 treatment-years for IDV versus 0.8/100 treatment-years for other PIs.91 In an Italian study of 555 patients taking IDV-based regimens, 24% had at least one episode of renal colic over 2 years and 50 (9%) stopped IDV.92
Crystals in the urine, most commonly composed of IDV base, occur in 32–67% of patients.93–96 particularly in the setting of increased urine pH.94,97 In vitro experiments found that IDV crystallizes in the loop of Henle98 and this may be promoted by the presence of RTV crystals.97 In one study, crystalluria occurred most commonly during the first 2 weeks after starting treatment and thereafter could be demonstrated in ∼25% of urine sediment samples.95 In another study of 54 patients starting IDV, 25% also had crystals seen on urinalysis.96 A urine pH < 6 was associated with few episodes of crystals, regardless of urine specific gravity, while nearly half of patients with a urine pH >6 and a urine specific gravity >1.015 had crystalluria.
Other urinalysis abnormalities that occur commonly in the setting of crystalluria are proteinuria, hemoglobinuria, and pyuria. Also described is a more chronic lesion associated with pyuria and characterized by diffuse interstitial nephritis with eosinophilia and scarring and tubule necrosis and dilatation with crystals99 and multinucleated histiocytes on urine cytology.100 Case reports have linked IDV crystalluria to a foreign body giant cell reaction with acute tubulointerstitial nephritis demonstrated on renal biopsy.101 Pyuria is common. In a Dutch cohort study of 184 patients taking IDV over a year, 35% developed pyuria at least once and of 134 evaluated, 24% had persistent pyuria.102 Patients may experience one of several other clinical syndromes. Back or flank pain with crystalluria without nephrolithiasis or dysuria and urgency with crystalluria.93 It is not clear if crystalluria always leads to nephrolithiasis.
Nephrolithiasis, diagnosed in the setting of flank pain with or without macroscopic or microscopic hematuria, was reported initially in 193 (9%) of 2071 patients from pooled clinical studies of IDV.1 A total of seven of 193 patients (4%) went on to discontinue the drug, whereas others resumed therapy. However, in one series of 155 patients, 43% developed nephrolithiasis over 78 weeks.103 In the ACTG 320 trial, the largest single clinical study of IDV, with 1156 patients randomized to zidovudine and lamivudine with or without IDV, seven patients (1%) experienced renal colic or nephrolithiasis (or both) of severe or greater intensity in the IDV group compared with no patients in the nucleoside analog group after a median follow-up of 38 weeks.104 In a smaller clinical study with 3 years of follow-up, 12 of 33 (36%) patients ultimately experienced clinical signs of nephrolithiasis105 and additional episodes occurred with continued therapy.106
Specific risks of stone formation that have been suggested include advanced age,102 female gender,107 concomitant hepatitis,108,109 and living in a warm climate or exercising (because of the risk of dehydration).110,111 Neither a prior or family history of kidney stones nor the concurrent use of acyclovir, sulfa drugs, or vitamin C has been definitively associated with developing nephrolithiasis while taking IDV.1 One group compared 15 evaluable patients taking IDV with urologic complaints and found that 14 of 15 (90%) had a higher IDV plasma concentration than the mean in a control group of 14 asymptomatic patients taking IDV.112 In six of these patients, the IDV dose was reduced to 600 mg q8h; plasma concentrations fell within the 95% confidence interval of the control group, and the patients remained asymptomatic. A high frequency of nephrolithiasis also has been reported in pediatric patients (29%).1
IDV nephrolithiasis may be difficult to demonstrate radiologically without the use of intravenous contrast dye because the stones are radiolucent.113 In general, crystalluria and kidney stones are not associated with changes in renal function, and they can be managed with analgesics, hydration, or interruption of the drug for 1–3 days. In one series, 11 (70%) of 18 episodes of IDV-induced nephrolithiasis were successfully managed with hydration and analgesia over the first 48 h.114 Others have recommended diuresis and acidification of the urine.115 Of 193 patients on IDV who developed nephrolithiasis, six (3%) had hydronephrosis and six (3%) underwent stent placement.1 However, hypertension, renal atrophy, renal failure, papillary necrosis, frank obstruction, and anuria also have been reported.93,116–120
Daudon and colleagues analyzed kidney stones passed from 29 referred patients taking IDV-containing regimens for periods of 1–20 weeks; they showed the stones to consist of crystals of IDV base monohydrate.121 Seven patients had stones also containing small amounts of calcium oxalate or calcium phosphate. It is of interest that the cores of all stones analyzed were made of IDV, strongly suggesting that IDV was the promoter of stone formation. Nephrolithiasis occurs more frequently at dose of IDV higher than 2.4 g/day122 or with IDV/RTV.1
In one retrospective analysis of 106 patients taking IDV, 19% experienced a sustained creatinine elevation of at least 20%, associated with low urinary specific gravity and pyuria, consistent with a crystal nephropathy.123 All abnormalities were reversible upon discontinuing the drug. In another retrospective analysis of 72 patients, the mean serum creatinine levels increased to more than 1.3 mg/dL in 13 (18%); this increase occurred more commonly in women and was associated with pyuria and microhematuria.107 Among 30 children taking IDV, 53% developed persistent sterile pyuria after 96 weeks and 33% had serum creatinine levels >50% above normal.124
BILIRUBIN AND HEPATIC TRANSAMINASE ELEVATIONS
Hyperbilirubinemia resulting from elevated indirect bilirubin (higher than 2.5 mg/dL) occurs in ∼10% of patients taking IDV in pooled studies and is dose-related.1 Unconjugated bilirubin is conjugated with glucuronic acid for biliary excretion. In an in vitro system, IDV inhibited uridine 59-diphospho-glucuronosyltransferase 1A1 (UGT1A1) activity, thereby decreasing bilirubin glucuronidation and likely explaining reversible hyperbilirubinemia.125 In a rat model, IDV competitively inhibited bilirubin UDP-glucuronosyltransferase (UGT) activity and increased plasma bilirubin levels.126 Others demonstrated IDV potently inhibited OATP1B1, a liver-specific organic anion uptake transporter, and postulated this as the mechanism for IDV-induced hyperbilirubinemia.127 Most commonly hyperbilirubinemia is subclinical, although occasionally mild scleral icterus occurs. Hyperbilirubinemia may resolve spontaneously and is reversible with discontinuation of the IDV. Fewer than 1% of patients have associated hepatic enzyme elevations.1
Patients with preexisting Gilbert’s syndrome may experience the highest bilirubin values. Swiss investigators studied 96 patients and characterized the contribution of the polymorphism conferring Gilbert’s syndrome (UGT1A1*28) and IDV on bilirubin levels.128 Overall, IDV increased bilirubin levels 0.46 mg/dL and 67% of individuals homozygous for the UGT1A1*28 polymorphism had at least two episodes of bilirubin >2.5 mg/dL vs 7% without the polymorphism. It is uncommon for a patient taking IDV to have a total bilirubin level higher than 5.0 mg/dL. In Merck studies 028 and 033, ∼7–8% of 302 patients receiving IDV had total serum bilirubin levels higher than 2.5 mg/dL.86 In the ACTG 320 trial, 27 of 577 subjects (5%) taking IDV had total bilirubin more than 2.5 times the upper limit of normal, and six others (1%) had total bilirubin more than five times normal.104
Laboratory abnormalities other than hyperbilirubinemia are less common. One group described eight (7%) of 117 patients who developed severe hepatitis (more than 5.0 times normal or 3.5 times baseline transaminase levels) after starting IDV, although this was similar to that for a group of patients taking nucleoside analogs only; there was a threefold higher risk in patients with chronic viral hepatitis.129 The same group followed 1161 patients taking PI-based regimen including 194 taking IDV (with or without RTV) and found 13% taking IDV had hepatic transaminases increased more than five times normal.130 Overall, patients with viral hepatitis accounted for 63% of these cases of hepatotoxicity, however more than 85% of patients with viral hepatitis had no hepatotoxicity.
METABOLIC DISORDERS
IDV was significantly associated with hyperglycemia (hazard ratio 4.0) compared to other antiretroviral drugs.131 The mechanism is thought to be that IDV is a relatively selective, noncompetitive inhibitor that binds one of the glucose transporters (GLUT4) involved in insulin-stimulated glucose uptake by adipocytes.132–134 Others have suggested that IDV may interfere with insulin signaling in some cell types, but does not impair insulin secretion.135–137 IDV acutely increases glucose and insulin levels in rats, and reduces glucose transport and insulin-induced GLUT4 activity.138,139 A single dose of IDV given to HIV negative volunteers decreased insulin-stimulated glucose transport.140 Two groups reported that 4-week courses of IDV in HIV-negative volunteers resulted in increased glucose production and decreased insulin sensitivity.141,142 Dube and colleagues reported 12 HIV-infected subjects who received 2 weeks of IDV monotherapy (and then added nucleoside reverse transcriptase inhibitors (NRTI) for 6 weeks) and found fasting glucose levels increased and insulin sensitivity decreased without a compensatory increase in insulin release.143
IDV, like most other HIV PIs, also has been associated with increased serum lipid levels. In the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) Study, a collaborative group of 11 cohorts following 11 205 patients, 7729 (43%) received a PI (including 2354 on IDV) and total cholesterol and triglyceride levels were highest in patients receiving RTV (with or without a second PI).144 The Swiss Cohort Study found lopinavir/RTV was associated with fewer lipid abnormalities compared to IDV/RTV.145 A meta analysis of three studies found no difference in triglyceride levels between groups taking dual NRTI with or without IDV.146 The mechanism of hyperlipidemia remains unknown, although it may be due to changes in serum lipoprotein lipase activity, hepatic lipid metabolism,147 or altered retinoid signaling148 (see Chapter 71).
A fat redistribution syndrome characterized by accumulation of abdominal fat and/or loss of peripheral fat with buffalo hump, lipomas, and in women, breast enlargement, has been associated with HIV PIs149–151 (see Chapter 72). The syndrome has been associated with increased serum lipid levels and insulin resistance150,151 and is thought to be due to inhibition of sterol-regulatory element-binding protein (SREBP)-1c, a regulator of cholesterol, triglyceride, insulin and adipocyte function and differentiation.152–154 The abdominal enlargement is characterized by an increase in visceral adipose tissue, typically without an increase in total body weight.149 During postmarketing surveillance of 282 reports of fat redistribution in patients taking IDV reported to Merck, 159 (56%) had fat accumulation, 60 (21%) had peripheral wasting, and 63 (22%) had both.155 Fat accumulation was more common in men and peripheral wasting more common in women. Weight gain was reported in 100% of those with fat accumulation, and weight loss was reported in 83% of those with peripheral wasting. In an Italian study, IDV exposure was significantly associated with developing both lipoaccumulation and lipoatrophy (RH 4.78 per year of exposure, p = 0.03).156 Morphologic changes typically do not resolve over the short term upon discontinuing IDV.150
Long-term sequelae of hyperlipidemia and fat redistribution are not known, although one group found no association between IDV therapy and myocardial infarction.157 Henry and colleagues studied 100 randomly selected patients taking an IDV-based combination regimen and found increased median levels of cholesterol (185 mg/dL, with 39% more than 200 mg/dL), triglycerides (184 mg/dL, with 12% more than 400 mg/dL), decreased median HDL levels (33 mg/dL), and a 56% rate of insulin resistance.158 They noted that these risk factors have been associated with an increased risk of coronary heart disease and warrant risk factor modification efforts. In addition, Shankar and colleagues studied eight HIV-negative men given IDV for 4 weeks and found impaired endothelial function159 and another group suggested IDV is associated with hypertension.116,160 However, Henry and colleagues also reported 79 patients taking IDV with suppressed HIV RNA levels with available pretreatment C-reactive protein (CRP) levels and in 56 followed for an average of 31 months found stable or decreased CRP levels.158 Young et al followed 11 HIV-infected patients starting IDV and found significant decreases in tissue plasminogen activator antigens, and the soluble inflammatory marker soluble TNF receptor (STNFr) and postulated that this was due to improvements in HIV-related inflammation.161
OTHER TOXICITIES
Gastrointestinal symptoms occur with IDV-containing regimens. In Merck studies 028 and 033 (IDV vs zidovudine vs the combination) of antiretroviral-naive patients, 4–12% of patients randomized to IDV alone reported abdominal pain, nausea, diarrhea, or vomiting of moderate or greater intensity.86 In the same studies, 4–14% of patients randomized to zidovudine reported these symptoms, as did 4–32% of patients randomized to the combination regimen. In studies of zidovudine-experienced patients, there were no differences in gastrointestinal side effects among patients taking a combination of zidovudine and lamivudine with or without IDV.104,162
The FDA issued a warning letter in July 1996 noting 15 case reports of spontaneous bleeding episodes in HIV positive hemophiliacs taking HIV PIs. In a follow-up report using the FDA’s postmarketing spontaneous reporting system, 39 of 67 (58%) reports of spontaneous bleeding in patients taking IDV occurred in hemophiliacs, compared to only two of 63 (3%) in hemophiliac patients taking zidovudine.163 Bleeding episodes tended to resolve upon discontinuation of therapy but recurred in two hemophiliac patients upon rechallenge. The mechanism is not known, though it may involve a direct effect on blood vessels.164
IDV also has been associated with skin, hair, and nail changes, possibly due to inducing gene expression of retinal dehydrogenase (RALDH), a key enzyme involved in retinoic acid synthesis.165 In one series of 101 patients who started an IDV-based combination regimen, 48 (57%) developed cheilitis, 34 (40%) dry skin with pruritus, five (6%) pyogenic granuloma of the toenails, and one (1%) severe alopecia.166 A localized skin rash was described in 110 patients who started IDV where 67% of the cohort reported onset of the rash within the first 2 weeks of starting therapy, 49 (44%) reported spread that involved the whole body, 86% had associated pruritus, and 81% required symptomatic treatment with antihistamines or corticosteroids.167 In this case series, 59% continued IDV therapy despite the presence of the rash. Stevens–Johnson syndrome is a rare complication of IDV.168 Paronychia have also been associated with IDV in case series169,170 with a fivefold increased risk associated with IDV treatment in one cohort study.171 In-grown toenails also have been described with IDV/RTV.172
TOXICITY OF IDV/RTV
In a study of 90 treatment-naive patients who received open-label therapy with two nucleoside analogs and IDV/RTV 400/400 mg twice daily, seven (8%) patients discontinued treatment over 24 weeks because of an adverse event: increased hepatic transaminases (n = 1), nausea (n = 4), and diarrhea (n = 2).173 Over the same time, 20% experienced nausea, 10% had circumoral paresthesias, 47% had a cholesterol level higher than 240 mg/dL, and 67% had a triglyceride levelhigher than 200 mg/dL. In a 48-week, open-label randomized study in which 106 patients received a combination regimen containing either the standard dose of IDV three times daily or IDV/RTV 800/100 mg twice daily, adverse events were evenly distributed except for nausea (48% IDV, 68% IDV/RTV; P = 0.04) and dry mouth (24% IDV, 46% IDV/RTV; P = 0.02).174 Solas and colleagues studied 63 patients taking IDV 800 mg/RTV 100 mg bid and linked an IDV trough level of >500 ng/mL to increased toxicity (P < 0.05).175 The incidence of nephrolithiasis is higher in patients receiving IDV/RTV compared to IDV.1
EFFICACY
Monotherapy and Dual Therapy
A 24-week randomized, blinded study (Merck 006) of IDV at either 200 or 400 mg q6h or zidovudine 200 mg q8h was conducted in 73 p24-antigenemic, mostly zidovudine-experienced HIV-positive patients.85,176 Of the 23 patients in the 400 mg dose cohort, 21 had greater than 1 log10 copies/mL decreases in HIV RNA at some point over 24 weeks compared with 12 of 21 in the 200-mg group and one of 29 in the zidovudine group. A dose-escalation study (Merck 021) evaluated IDV at doses of 800 mg q8h, 1000 mg q8h, and 800 mg q6h in 70 HIV-positive patients.22 Over 24 weeks the HIV RNA levels decreased ∼2 log10 copies/mL, without differences among the three treatment groups.
A large Phase II study (Merck 028) compared IDV 800 mg q8h, zidovudine 200 mg q8h, or a combination of the two in 996 antiretroviral therapy-naive patients with baseline CD4-lymphocyte counts of 50–250/mm3.86 During the study the protocol was amended to add lamivudine to the zidovudine-containing arms. A protocol-defined interim analysis of the study found highly significant reductions in clinical progression in the IDV arms compared to the zidovudine arm; and the study was terminated early. Over a median follow-up of 1 year, reductions in the hazards of clinical progression of 70% (combination group) and 61% (IDV group) were seen over that in the zidovudine group (P < 0.0001). In addition, significant changes in HIV RNA and CD4-lymphocyte counts were seen in the IDV groups compared to the zidovudine group.
THREE-DRUG COMBINATION: IDV AND TWO NRTIs
A three-drug combination study (Merck 035) enrolled 97 patients with zidovudine experience without prior lamivudine or PI experience who had an HIV RNA level of 20 000 copies/mL or more. They were randomized to IDV 800 mg q8h, zidovudine 200 mg q8h/lamivudine 150 mg bid, or a combination of all three drugs.162 At baseline, patients had taken zidovudine for a median of 31 months and had a median HIV RNA level of 43 200 copies/mL and a median CD4-lymphocyte count of 144/mm3. At 24 weeks of follow-up, the median HIV RNA changes were −1.2 log10 copies/mL in the IDV group, −0.8 log10 copies/mL in the zidovudine/lamivudine group, and −1.8 log10 copies/mL in the triple combination group. At the same time, 43% of the IDV group decreased HIV RNA to less than 500 copies/mL compared with 0% of the zidovudine/lamivudine group and 90% of the triple-combination group. Most patients on triple therapy who had viral loads of less than 500 copies/mL also had levels of less than 50 copies/mL using an ultrasensitive viral load assay.
After at least 24 weeks of original therapy, all patients received triple-combination therapy and continued to be followed. The simultaneous introduction of three-drug antiretroviral therapy achieved a virologic effect superior to sequential introduction of the same three drugs.177 Results with the triple combination were durable in most patients through 6 years of follow-up, with 53% and 47% of patients suppressing HIV RNA levels to less than 500 and less than 50 copies/mL, respectively, using an intent-to-treat analysis.106 At the same time, the CD4-lymphocyte count increased +268 cells/mm3 over baseline (Fig. 17-2).
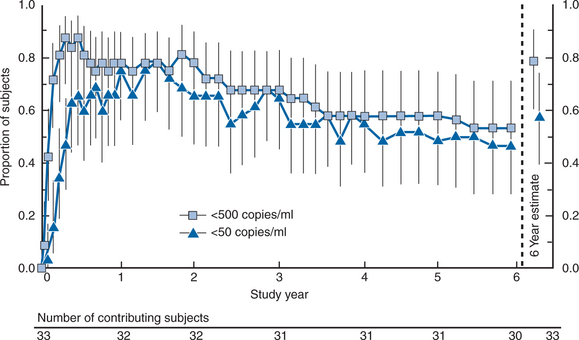
Reprinted from Gulick RM, Meibohm A, Havlir D, et al. Six-year follow-up of HIV-1-infected adults in a clinical trial of antiretroviral therapy with indinavir, zidovudine, and lamivudine. AIDS 17:2345, 2003.106
A parallel study (Merck 039) enrolled 320 patients with zidovudine experience without prior lamivudine or PI experience who had 50 CD4+ T-lymphocyte cells/mm3 or less and randomized them to IDV monotherapy, zidovudine/lamivudine, or the triple combination.178 At baseline the mean HIV RNA was 74 353 copies/mL and the CD4+ T-lymphocyte count was 18/mm3. The mean HIV RNA changes from baseline at 24 weeks were −0.36 log10 copies/mL in the IDV group, −0.25 log10 copies/mL in the zidovudine/lamivudine group, and −1.93 log10 copies/mL in the three-drug combination group. The proportion of patients with HIV RNA levels of less than 500 and less than 50 copies/mL were 3% and 2% in the IDV group, 0% and 0% in the zidovudine/lamivudine group, and 56% and 45% in the triple-drug combination group, respectively. In the 3-drug group, these results were updated through 4.5 years of follow-up with 24% (intent-to-treat) and 92% (observed) with HIV RNA levels <500 copies/mL and 22% (intent-to-treat) and 85% (observed) with HIV RNA levels <50 copies/mL.179
A large study of similar combination antiretroviral regimens (ACTG 320) assessed clinical endpoints.104 In this study, 1156 patients with CD4+ T-lymphocyte counts of 200/mm3 or less (median 86/mm3) with zidovudine experience but without previous lamivudine or PI use were randomized to zidovudine 200 mg q8h/lamivudine 150 mg bid with or without IDV 800 mg q8h and were followed for the occurrence of a new AIDS-defining illness or death. After a median follow-up of 38 weeks, the Data and Safety Monitoring Board (DSMB) recommended stopping the study because of a significant difference between the two treatment arms: 63 patients (11%) taking the two-drug combination had a new AIDS event or death versus 33 patients (6%) on the triple-combination arm (P = 0.001) (Fig. 17-3). The three-drug combination of IDV/zidovudine/lamivudine demonstrated a 50% decrease in the rate of AIDS or death over the zidovudine/lamivudine regimen. A subanalysis also found a significant improvement in health-related quality of life after 24 weeks, particularly in patients with low baseline CD4+ T-lymphocyte counts.180
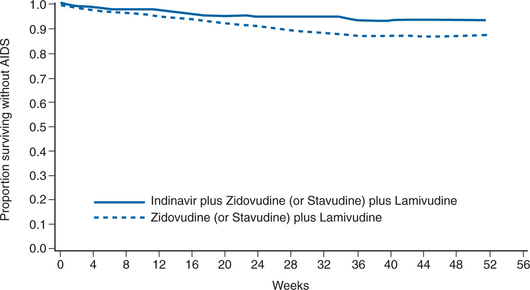
From Hammer SM, Squires KE, Hughes MD, et al. A randomized placebo-controlled trial of indinavir in combination with two nucleoside analogs in human immunodeficiency virus infected persons with CD4 cell counts less than or equal to 200 per cubic millimeter. N Engl J Med 337:725, 1997; Copyright 1997, Massachusetts Medical Society.104 All rights reserved.




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


