Fig. 5.1
(Right side) Healthy periodontal tissue. (Left half side) Effects of periodontitis with subgingival bacterial plaque formation, gingival tissue inflammation, periodontal attachment loss, periodontal pocket formation, and alveolar bone loss
Pathogenic Mechanisms
The chronic inflammatory condition of periodontitis is induced by pathogenic biofilm or dental plaque, which accumulates on the tooth surface. Over 500 bacterial species have been detected in dental plaque; however, the composition of the causative bacterial species is still debated [25–28]. The classic periodontal pathogens are Gram-negative bacteria such as Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticola [29]. Recent studies have identified other bacteria that may be pathogenic [30]. Although bacteria are necessary for periodontal disease to take place, a susceptible host is also needed [27]. The predisposing factors that make an individual susceptible have not yet been defined though periodontal disease increases considerably in aged humans and animals including dogs and mice and by conditions that increase inflammation such as diabetes [31]. The inflammatory process occurring in periodontitis is characterized by the infiltration of leukocytes, which limit the level of bacterial invasion and at the same time may be harmful to the periodontal tissue [31]. The destruction of periodontal ligament and bone is thought to be the result of a disruption of the homeostatic balance between the host response and bacteria that result in inflammation in close proximity to bone [31–33]. The host immune response to bacteria or their products stimulates production of osteoclastogenic factors by immune cells and cells of osteoblastic lineage, which then induce bone loss. Periodontal disease in humans and in experimental animal models is linked to both the innate and adaptive immune response. Several studies have reported that individuals with periodontitis exhibit increased levels of interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) in gingiva and crevicular fluid in the gingival sulcus [34]. Both genetic deletion and specific inhibition of these cytokines have been found to reduce periodontal disease progression [34–37]. Similarly, mediators of the adaptive immune response are elevated in individuals with periodontal disease and inhibition or genetic deletion of mediators such as receptor activator of nuclear factor kappa-B ligand (RANKL) and interferon-γ (IFN-γ) result in reduced periodontal disease progression [36, 38, 39]. Our laboratory has recently shown that the production of factors by osteoblasts and osteocytes also contributes to osteoclast formation and activity in periodontal disease (unpublished data). Evidence that the host response plays a critical role was also shown when treatment with a prostaglandin inhibitor reduced periodontitis-related bone loss [40] or inhibition of inflammatory cytokines such as IL-1 and TNFα [35, 37]. Thus, periodontitis is a complex disease, where multiple causal risk factors play simultaneous and interactive roles. Periodontal bone loss is affected by the bacterial biofilm which forms, ability of bacteria or their products to pass through the epithelial barrier into connective tissue, the status of the host response, and the presence of environmental stressors and/or systemic disease such as diabetes [41–43].
Impact of Diabetes on Periodontal Disease
Diabetes and periodontitis are two chronic diseases that are biologically linked [44, 45]. Periodontitis is one of the first clinical manifestations of diabetes [19]. Diabetes is an important risk factor for periodontitis [4, 46]. The risk of periodontitis is increased approximately 2–4 times in diabetic versus nondiabetic subjects [4, 47]. In one study, periodontitis was found in 60 % of T1DM patients compared to 15 % without diabetes [48]. Patients with diabetes are at higher risk of severe periodontitis compared with nondiabetic subjects [49]. A study in African Americans found 70 % T2DM patients had moderate periodontitis and 29 % had severe disease, which is significantly higher than the prevalence of 11 % without diabetes [50]. The severity of periodontitis is directly proportional to the level of glucose control [48, 51].
Impact of Diabetes on Gingiva/Gingivitis
Periodontitis is preceded by various stages of gingival inflammation referred to as gingivitis. Gingivitis in T1DM children and adolescents is twice that of matched control subjects [52]. Similarly, higher rates of gingival inflammation occur in T2DM adults compared to adults without diabetes. Nearly, 64 % of patients with T2DM have gingivitis compared with 50 % of subjects without diabetes [52]. The degree of metabolic control of diabetes is an important factor in the development and progression of gingivitis; good metabolic control significantly reduces gingivitis [53, 54].
Diabetes may affect periodontal disease through a number of different avenues including an impaired antibacterial defense. However, studies investigating whether diabetes causes a change in oral flora have had inconsistent results and the issue has not yet been settled. An alternative explanation is that diabetes alters the inflammatory response to periodontal pathogens. This is supported by studies examining the response to a well-defined inoculum of periodopathic bacteria [55]. Under normal circumstances, inflammation normally resolves through an active process regulated by cellular signals [56, 57]. However, resolution of the inflammation is impaired in the diabetic animals [58]. Thus, one explanation for enhanced risk of periodontal disease in diabetic individuals is the presence of a greater inflammatory response to bacteria on the tooth surface that stimulates the formation of osteoclasts and suppresses the repair process [55, 59].
In animal models, diabetes leads to increased production of TNF in the epithelium and connective tissue [60]. Periodontal infection causes an increase in apoptosis of gingival epithelial cells, fibroblasts, and bone lining cells that is significantly enhanced by diabetes in a caspase-3-dependent mechanism [60]. This may be important because diabetes-enhanced inflammation and apoptosis negatively impact the gingiva by causing a loss of epithelial barrier function and inhibiting repair processes [61, 62]. High levels of TNF-α can stimulate the expression of pro-apoptotic genes in diabetics [58, 63].
Impact of Diabetes on Periodontal Ligament/Loss of Attachment
The gingiva is attached to the root surface of teeth by collagen bundles that integrate with the tooth surface. Loss of this attachment is one of the hallmarks of periodontitis and occurs in conjunction with bone loss. Diabetes increases attachment loss, which is worsened by poor glycemic control [48, 64]. More than 25 % of T1DM patients with poor metabolic control have sites with moderate to severe signs of periodontitis compared to only 10 % of subjects with good metabolic control [52]. Moreover, the severity is proportional to the duration of diabetes [51, 65].
Impact of Diabetes on Alveolar Bone Loss
Periodontitis is the most common osteolytic disease in humans and is aggravated by diabetes [66]. Diabetes potentiates the severity of periodontitis and accelerates bone resorption. The number of sites with bone loss in poorly controlled T1DM individuals is twice that of nondiabetic subjects [67]. Animal studies demonstrate that periodontitis is increased threefold in T1DM rats compared to normal rats [68] and is significantly higher in T2DM rats [61]. The risk and degree of alveolar bone loss is positively correlated with the lack of metabolic control [69].
Effect of Diabetes on Osteoclasts in Periodontitis
Bone remodeling begins with the activity of osteoclasts, followed by new bone formation through the activity of osteoblasts. Under physiological conditions, the two activities are coupled, but the two processes are uncoupled in pathologic processes. Human studies generally indicate that diabetes mellitus increases osteoclastogenesis. Individuals with DM generally have increased systemic levels of bone resorption markers as indicated by higher circulating levels of tartrate-resistant acid phosphatase [70]. Animal studies show similar results [71]. T2DM rats have increased osteoclastic bone resorption in periodontal bone compared to normoglycemic controls [61]. Increased inflammation, ROS, and AGEs are thought to increase osteoclast activity.
Increased Inflammation Activate Osteoclasts in Diabetes
Diabetes has been shown to enhance osteoclast formation in inflammatory areas. Type 2 diabetic rats have a ~2–4-fold increase of the osteoclast number after bacterial infection by oral inoculation of a periodontal pathogen or ligature-induced periodontitis compared with control rats [59–61]. T1DM with periodontitis also have a 2–4-fold increase in the number of osteoclasts compared to nondiabetic rats with periodontitis [72]. A higher degree of inflammation and a more persistent inflammatory response following periodontitis [55, 73] may lead to greater stimuli for osteoclastogenesis.
In diabetic mice, TNF-α, macrophage colony-stimulating factor, RANKL, and vascular endothelial growth factor-A (VEGF-A) are up-regulated which can directly promote osteoclast differentiation and activation [74, 75]. Diabetes increases TNF levels that has been shown to prevent downregulation of genes associated with host defense, apoptosis, cell signaling and activity, and coagulation/hemostasis/complement [58]. Similarly, patients with periodontitis and diabetes have significantly higher levels of IL-1β, TNF-α, and prostaglandin E2, which result in more prolonged osteoclast formation and activity [76, 77]. Enhancement of IL-17, IL-23 in periodontitis in type 1 diabetic subjects, and overexpression of IL-1β, IL-6 in type 2 diabetic patients have been reported, which result in enhanced osteoclastogenesis and prolonged duration of inflammatory responses [78, 79]. T2DM patients with periodontal disease have increased levels of TNF-α and IL-6, which was also associated with increased dyslipidemia and lipid peroxidation [80]. Increased fatty acid levels in diabetes mellitus may also enhance osteoclastogenesis [81]. In addition to increasing inflammation, diabetes also impairs the resolution of periodontal inflammation. The importance of resolving inflammation has been demonstrated by treatment of animals with periodontitis with resolvins [82] or by treatment of diabetic animals with TNF inhibitors [58, 59].
Increased ROS Activate Osteoclasts in Diabetes
High levels of ROS contribute to diabetes-related periodontitis. Invading bacteria trigger the release of cytokines and chemokines that induce neutrophil recruitment and activity and which subsequently release ROS in periodontal tissues [83, 84]. Neutrophils from diabetic patients produce more superoxide than neutrophils from normal subjects [14]. The imbalance between production of ROS and antioxidant defense results in increased oxidative stress [85]. The formation of AGEs also increases oxidative stress in the periodontal tissues. It has been shown that ROS such as superoxide and hydrogen peroxide activate osteoclasts and promote osteoclast formation [86]. A related process, lipid peroxidation, is also linked to increased periodontal disease T2DM and a greater inflammatory response in the periodontal tissues in humans [80, 87]. Patients with T2DM show elevated mitochondrial ROS which promotes RANKL-mediated osteoclast differentiation and function [88].
Increased AGEs Activate Osteoclasts in Diabetes
In vitro studies suggest that hyperglycemia predisposes to increased osteoclast formation [89]. AGEs also increase osteoclasts activity [90]. Osteoclast-like cells express RAGE, which serves as a positive factor to regulate the osteoclast formation [91]. Mice that lack the RAGE have increased bone mass and decreased osteoclast numbers compared to wild-type mice [91], supporting the concept that AGEs contribute to osteoclast formation in diabetes. Diabetes enhances the formation of AGEs in the periodontium and increases expression of RAGE [92]. The level of AGEs in the gingiva is increased in both type 1 and type 2 diabetes-associated periodontitis [93]. It has been shown that AGE–RAGE interaction on monocytes activates the transcription factor NF-κB, which alters the phenotype of the monocyte/macrophage and results in the increased production of proinflammatory cytokines [94]. RAGE stimulation may contribute to osteoclastogenesis via increased expression of receptor activator of RANKL and downregulation of osteoprotegerin (OPG) [95].
RANKL interacts with its receptor on the surface of osteoclast precursors to induce osteoclast formation and activity. OPG inhibits osteoclast formation by binding to RANKL [96]. A number of studies focusing on osteoclastogenesis-related factors have reported elevated expression of RANKL and TNF in diabetes-associated periodontal tissues [59, 96]. Studies with animals suggest that RANKL/OPG ratios and the level of other inflammatory cytokines such as TNF are critical mediators for the enhanced osteoclastogenesis in diabetes in periodontal disease [59, 97]. TNF levels and high RANKL/OPG ratios in the periodontium in humans are negatively influenced by poor glycemic control in subjects with diabetes [80, 98]
Effect of Diabetes on Osteoblasts in Periodontitis
Bone resorption is followed by a period of bone formation, a coupling process that limits the amount of net bone loss, which occurs during the resolution of inflammation in the periodontium [20]. We found that T2DM rats do not generate a burst of bone formation that normally occurs following induction of periodontal disease [61]. Inflammation plays an important role in this effect by limiting repair of resorbed bone. This occurs by reducing osteoblast numbers through decreased proliferation of precursors and greater apoptosis of mature osteoblasts [59, 63]. These studies suggest a molecular basis for the negative impact of T2DM on bone by the effect of diabetes-enhanced inflammation on suppressing the expression of factors such as fibroblast growth factor or bone morphogenetic proteins that are needed for new bone formation.
Diabetes Inhibits Osteoblasts Differentiation and Function
Diabetes also interferes with the bone formation by reducing the expression of transcription factors that regulate osteoblast differentiation [99]. In T1DM and T2DM rats, osteoblasts exhibit lower alkaline phosphatase activity and mineralized matrix formation [100, 101]. Inflammation has a significant effect on bone [102, 103]. Inflammation impairs the function of bone-forming osteoblasts by suppressing mature osteoblast function such as the production of bone matrix [104]. One of the striking features of diabetes is elevated levels of inflammatory mediators, particularly TNF [76]. Diabetic animals have higher levels of TNF in bone, which is associated with reduced bone formation and repair. TNF blocks the differentiation of osteoblasts, where inflammation is thought to be present [105]. This is consistent with reports that TNF inhibits differentiation of osteoblasts in vitro [106, 107] and also interferes with bone morphogenetic protein signaling [108].
Periodontal infection-induced alveolar bone loss in diabetic subjects is accompanied by enhanced expression of RAGE and production of AGEs in the gingival tissue [109]. AGEs have been shown to interfere with osteoblast differentiation and induce apoptosis of osteoblasts in diabetes via the mitogen-activated protein kinase and cytosolic apoptotic pathway [110]. When AGEs are applied to osseous wounds in normal animals, the rate of healing is reduced in half, indicating that AGEs, which are elevated in diabetes, contribute to impaired bone formation [111]. In addition, RAGE is expressed at higher levels in osteoblasts in diabetic conditions rendering diabetic animals even more sensitive to the effects of AGEs [111].
Mesenchymal stem cells (MSCs) represent a precursor pool of osteoblasts that are bone-forming cells. Inflammation, which is elevated in diabetic bone healing [112], has a significant effect on reducing MSC differentiation [113]. A mechanism through which inflammation affects MSC is through induction of NF-κB activation. Increased NF-κB activity interferes with wnt-stimulated MSC differentiation by increasing beta-catenin degradation [102]. Moreover, TNF suppresses activation of the Osx promoter [114]. This interferes with MSC differentiation to osteoblasts since osterix is needed in early steps of differentiation. AGEs also inhibit MSC differentiation. One mechanism involves AGE-induced up-regulation of ROS in MSCs that leads to reduced MSC differentiation [115, 116]. In human MSCs and mouse stromal ST2 cells, AGEs suppress osteogenic differentiation [117]. T2DM mice have fewer MSCs and these MSCs appear to have poor homing capability to injury sites [118]. T1DM rats have more numerous apoptotic cells in the bone marrow, and the size of osteoprogenitor pool is significantly reduced [101]. Thus, diabetes reduces the number of progenitors and inhibits differentiation of MSC to osteoblasts.
Diabetes Promote Osteoblast Apoptosis
Apoptosis of osteoblasts is significantly increased by diabetes. Diabetes leads to the up-regulation of pro-apoptotic mediators including TNF-α, AGEs, and the formation of ROS [119]. TNF can induce apoptosis by binding to the TNF receptor-1, which triggers the initial events in apoptosis [120]. Some of the detrimental effects of diabetes-enhanced TNF-α levels may be due to the induction of cell death by triggering caspase activity. Caspases are a family of cysteine proteases that can act as either initiators (caspase-2, 8, and 9) or executioners (caspase-3, 6, and 7) of apoptosis [121]. Caspase-3 appears to play a central role in bacteria and lipopolysaccharide (LPS)-mediated apoptosis [122, 123]. Additionally, TNF stimulates the expression of several pro-apoptotic genes, many of which are regulated by the pro-apoptotic transcription factor, forkhead box-O1 (FOXO1) [124]. There is evidence that both diabetes and bacterial infection in periodontitis enhances apoptosis of osteoblastic cells to reduce osseous coupling [63, 125], which may involve stimulatory signals from both the innate and adaptive immune response [112, 126]. CML-collagen, one of the AGEs found in bone, stimulates apoptosis of bone-lining cells in vivo and in various osteoblastic cell cultures mediated through RAGE [127] via the MAP kinase pathway [110].
The production of ROS is another mechanism of diabetes increasing apoptosis. Persistent inflammation and hyperglycemia leads to the cellular accumulation of ROS, which is linked to diabetic complications [66, 128]. Increased oxidative stress in periodontal tissue has been shown to lead to greater osteoblast apoptosis [129] and involve activation of caspase-3 [130].
Diabetes also increases loss of cells in the periodontal ligament from periodontal infection [61, 131]. This is significant since the periodontal ligament is a rich source of cells capable of differentiating into osteoblasts. Studies in diabetic animals indicate that diabetes causes a more than twofold induction of genes that regulate the apoptosis of osteoblasts and fibroblasts following bacterial infection and a fivefold increase in osteoblast apoptosis [122, 132].
Blocking apoptosis by treatment of diabetic rats with a caspase-3 inhibitor significantly increases the number of osteoblasts, which in turn leads to significantly greater amounts of new bone formation. Furthermore, the number of osteoclasts and their activity is increased by treatment with a caspase-3 inhibitor with the net effect increasing bone formation due to greater bone coupling. This is consistent with previous findings that a pancaspase inhibitor reduces apoptosis and increases new bone formation following bacterial infection [122]. Taken together the results indicate that bacterial infection in diabetic animals has a significant impact on periodontal disease through enhanced apoptosis of osteoblasts or their precursors.
Conclusion
In summary, individuals with diabetes mellitus have increased risk and severity of periodontal disease [9, 19, 133]. Periodontitis is one of the first clinical manifestations of diabetes [19]. Diabetes aggravates periodontitis by an increase in the inflammatory response to bacterial infection and reducing the capacity to downregulate inflammation [9, 55]. There is a direct link between persistent hyperglycemia, an exaggerated inflammatory response to periodontal pathogens and periodontal bone loss [31, 66]. The impact of diabetes on the periodontium involves inflammation associated with both the innate and adaptive immune response [8, 31]. Diabetes-enhanced inflammation increases osteoclastogenesis and decreases reparative bone formation. A number of factors are increased by diabetes including RANKL, AGEs, ROS, and TNF that stimulate osteoclasts. Moreover, diabetes prolongs inflammation leading to longer periods on osteoclast activity as well as interfering with subsequent bone coupling. The negative effect of inflammation on bone coupling is likely to be an important factor in the disease process [59, 112] as inflammatory cytokines interferes with bone morphogenetic protein and Wnt signaling and also stimulates osteoblast apoptosis [104, 108, 134, 135]. The mechanisms by which diabetes affects periodontitis are summarized in Fig. 5.2.
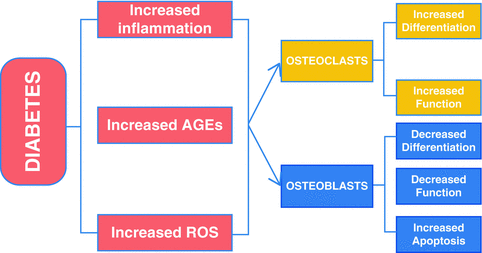
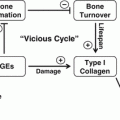
Fig. 5.2
Impact of diabetic mellitus on periodontitis. Diabetes increases inflammation (increased cytokine production such as TNFα), and the production of AGEs and ROS that can affect both osteoclasts and osteoblasts. These factors may increase osteoclast formation and activity and inhibit osteoblast differentiation, activity, and survival
References
1.
Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta-cell function. Nature. 2001;414(6865):788–91.PubMed
2.
Kahn SE. Clinical review 135: the importance of beta-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab. 2001;86(9):4047–58.PubMed
3.
Cavaghan MK, Ehrmann DA, Polonsky KS. Interactions between insulin resistance and insulin secretion in the development of glucose intolerance. J Clin Invest. 2000;106(3):329–33.PubMedCentralPubMed
4.
Preshaw PM, Bissett SM. Periodontitis: oral complication of diabetes. Endocrinol Metab Clin North Am. 2013;42(4):849–67.PubMed
5.
Cruz NG, Sousa LP, Sousa MO, Pietrani NT, Fernandes AP, Gomes KB. The linkage between inflammation and type 2 diabetes mellitus. Diabetes Res Clin Pract. 2013;99(2):85–92.PubMed
6.
Johnson DR, O’Connor JC, Satpathy A, Freund GG. Cytokines in type 2 diabetes. Vitam Horm. 2006;74:405–41.PubMed
7.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91.PubMed
8.
Nikolajczyk BS, Jagannathan-Bogdan M, Shin H, Gyurko R. State of the union between metabolism and the immune system in type 2 diabetes. Genes Immun. 2011;12(4):239–50.PubMed
9.
Graves DT, Kayal RA. Diabetic complications and dysregulated innate immunity. Front Biosci. 2008;13:1227–39.PubMedCentralPubMed
10.
Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058–70.PubMedCentralPubMed
11.
Pitocco D, Zaccardi F, Di Stasio E, Romitelli F, Santini SA, Zuppi C, et al. Oxidative stress, nitric oxide, and diabetes. Rev Diabet Stud. 2010;7(1):15–25.PubMedCentralPubMed
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


