Vaccination and adoptive immunotherapy for herpes virus infections has become an attractive option for the control of a virus family that negatively affects transplantation. In the future, enhanced ability to select antigen-specific T cells without significant in vitro manipulation should provide new opportunities for refining and enhancing adoptive immunotherapeutic approaches. This article focuses on advances in the area of vaccinology for some of these infections and in the use of adoptive immunotherapy. At present, many of these approaches in transplant recipients have focused on infections such as human cytomegalovirus, but the opportunity to use these examples as proof of concept for other infections is discussed.
Despite the progress in immunosuppressive drugs to manage and minimize organ rejection after transplantation, infectious complications remain a major cause of morbidity. Infections such as herpes simplex virus 1 (HSV-1) and varicella-zoster virus (VZV) have been successfully controlled through the deployment of prophylactic acyclovir or its prodrug valaciclovir, whereas prophylactic and preemptive therapy with valganciclovir has been instrumental in managing human cytomegalovirus (HCMV) infections. However, the management of Epstein-Barr virus (EBV), human herpes virus-6 (HHV-6), adenovirus, hepatitis C virus (HCV), hepatitis B virus (HBV), papillomavirus, and BK virus infections remains challenging. In addition, the side effect profile of ganciclovir and the possibility that drug resistance may develop with long-term prophylactic use in high-risk patients, such as those needing lung transplants, have stimulated interest in other management strategies. This article focuses on advances in the area of vaccinology for some of these infections and in the use of adoptive immunotherapy. At present, many of these approaches in transplant recipients have focused on infections such as HCMV, but the opportunity to using these examples as proof of concept for other infections is discussed.
Vaccination
HCMV Vaccines
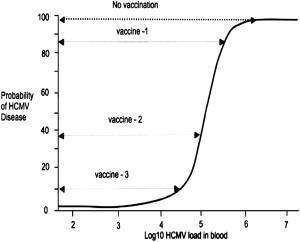
The earliest vaccine deployed for HCMV infections was the low-passage, live, attenuated Towne strain of HCMV by Plotkin and colleagues. The results of a trial in kidney transplant recipients showed that immunization was unable to prevent infection but was associated with a reduction in the severity of HCMV disease. Despite these encouraging findings, progress on an HCMV vaccine has been slow in the intervening years, with several vaccine preparations being formulated but, until recently, few showing sufficient protection to warrant further study. These vaccine preparations are summarized in Table 1 . A fundamental question relating to the use of vaccination to control and potentially eradicate HCMV infection is the level of vaccine coverage required to induce herd immunity in the general population, and of the vaccine efficacy required to control replication and disease after transplantation. A key study by Griffiths and colleagues showed that, in high resource countries such as the United Kingdom and United States where HCMV seroprevalence rates are approximately 60%, the basic reproductive number (Ro; the number of new infections arising from 1 infected individual) for HCMV in women of child-bearing age was approximately 2.4. Thus, a vaccine coverage of around 62% would be sufficient to lead to herd immunity and eradication of HCMV without a significant effect on the incidence of congenital HCMV infection/disease. In the transplant setting, vaccination could be deployed in high-risk patients who are seronegative or as a prophylactic vaccine in patients who are already seropositive for HCMV. One study provided an estimate of the basic reproductive number for HCMV in these 2 clinical settings for liver transplant recipients. These data indicate that in the D+R− setting, the Ro for HCMV is approximately 15, meaning that a vaccine would need to have an efficacy against replication of approximately 93% if it was to fully control replication. In contrast, the Ro for HCMV in the D+R+ group was reduced to 2.4, indicating that an HCMV vaccine deployed as an immunotherapeutic in these patients would only need to achieve an efficacy of about 60% to successfully control HCMV replication. The ability to affect the probability of HCMV disease is intimately linked to the relationship between viral load and disease. In all cases to date, viral load and probability of HCMV disease follow a sigmoidal pattern whereby the risk of HCMV disease increases substantially at certain viral-load thresholds. In the context of vaccination, a vaccine that did not eradicate infection/replication but which induced a sufficient level of immunity to partially control HCMV replication could still have a profound effect on the incidence and severity of HCMV disease. This concept is summarized in Fig. 1 and is consistent with the findings of the Towne vaccine study described earlier.
| Vaccine | Description | Status |
|---|---|---|
| Towne | Live attenuated | Being used in prime boost with DNA vaccines |
| gB | Recombinant, soluble | Phase II studies complete in healthy; undergoing phase I in solid-organ transplant recipients |
| Canarypox pp65 | Live single-cycle expression | Phase I |
| pp65, gB | DNA plasmid | Phase I |
| AlphaVax | Alphavirus expressing gB and pp65-IE1 fusion protein Single-cycle expression | Phase I |
At present, the most encouraging results for an HCMV vaccine have come from the recombinant glycoprotein B (gB) vaccine. gB is a major site for neutralizing antibodies and can adsorb up to 80% of the serum-neutralizing antibodies in healthy seropositive individuals. In addition, purified and recombinant gB has been shown to elicit high-level humoral immunity in animal models and protect against fetal loss in a guinea pig model. Recombinant gB consisting of the extracellular domain and the intracellular domain, but with a truncated transmembraneous domain, has been expressed in mammalian cell culture and subjected to phase I and II clinical trials. The vaccine is highly immunogenic when administered with the adjuvant MF59 (3 doses at months 0, 1, and 6) yielding neutralizing titers in seronegative volunteers that were in excess of those found with natural immunity. In addition, the vaccine boosted neutralizing titers when given to seropositive individuals. The results of a randomized double-blind, placebo-controlled, phase II study in 464 seronegative women within 1 year of them giving birth have recently been reported. The vaccine efficacy was 50%, reducing the number of seroconversions from 31 in the placebo group to 18 in the vaccine group. These results are encouraging and have rekindled interest in the HCMV vaccine area by small and large pharma.
Do these results have any effect on the vaccination of transplant patients against HCMV? One of the challenges in the transplant setting is the need for the gB vaccine to be given as 3 doses over a period of 6 months. Many patients proceed to transplant before the full course of vaccination can be given, and so the likelihood of protection may be reduced. Nevertheless, one study ongoing at the Royal Free Hospital, London, and coordinated by scientists at University College London, has recruited renal and liver transplant patients (seropositive and seronegative before transplant) into a placebo-controlled trial of the gB-MF59 vaccine with an end point of viral replication (incidence and kinetics) after transplantation. At the time of writing, this trial has not reported its results, but these should be available in 2010.
Human Papillomavirus Vaccines
Papillomavirus infections remain an important pathogen in immunocompromised men and women with HIV or in transplant recipients, for whom the risks for common warts and anogenital lesions attributable to human papillomavirus (HPV) are substantially increased compared with healthy individuals. In particular, female renal transplant recipients have a substantially increased risk for HPV-associated anogenital cancers and cervical intraepithelial neoplasia. The development of HPV-related anal tumors has also been documented after liver transplantation in a retrospective analysis. Anal HPV-DNA was detectable in 23% of men and women before the induction of immunosuppression for a liver or kidney transplant. The recent licensing of recombinant HPV vaccines based on the L1 protein of HPV types 6, 11, 16, and 18 for the prevention of genital warts, cervical dysplasia, and cervical cancer has now provided opportunities to deploy such vaccines in the transplant setting. Because these vaccines work best in individuals before HPV exposure, it is likely that the first deployment may occur in the pediatric transplant setting. In the setting of immunosuppression, the safety of HPV vaccines has yet to be determined. However, extrapolation from the use of other nonlive vaccines suggests that HPV vaccine would be safe and induce effective, if diminished, immune responses, with no associated graft rejection or other serious adverse effects. In addition, given the risks of anogenital lesions, the ability to boost preexisting immunity to reduce or prevent anogenital lesions is also likely to be an important consideration in adult populations, especially in women receiving renal transplants. However, it is interesting that a recent cost-effectiveness analysis suggests that a program of HPV vaccination before transplantation is unlikely to be cost-effective. Nothwithstanding this, clinical trials of HPV vaccines are underway in centers in North America.
VZV Vaccine
To date, a prophylactic vaccine against VZV is the only live, attenuated herpes virus vaccine to be licensed in any country. It was initially tested, and has been widely used for the past few decades, in Japan. The currently available vaccine consists of attenuated virus from the Oka strain of VZV and has been in use safely and successfully in healthy children and adults in many other parts of the world.
Among immunocompromised patients, including transplant recipients, varicella infections may be severe and even fatal. The clinical presentation may be atypical in immunocompromised patients, in whom the rash may be nonvesicular or even absent. Immunocompromised children and adults may present with abdominal zoster, characterized by abdominal and back pain, which may precede or occur in the absence of skin lesions, posing a diagnostic challenge. VZV-associated cerebral vasculitis has also been described in the setting of immunocompromised patients. In addition, recovery from VZV infection may be slow, with individuals remaining infectious for up to several weeks following initial clinical presentation. It is therefore important to consider protection of these susceptible populations with the use of the VZV vaccine, 2 doses of which provide 75% protection against any disease and at least 95% protection against severe disease and its complications among immunocompetent adults.
The use of the live, attenuated varicella vaccine has been most extensively studied in children with acute lymphoblastic leukemia (ALL), but studies involving other immunocompromised populations are limited. In a multicenter trial, the vaccine was shown to be safe and immunogenic when administered to children with ALL who had suspended maintenance chemotherapy for at least 1 week before and 1 week after vaccination. Seroconversion rates varied from 42% to 96% and were, on average, lower than in healthy children. Incidence of recurrent varicella (herpes zoster) was also found to be less in immunocompromised vaccine recipients compared with that observed following natural infection. In children with asymptomatic or mild symptoms of HIV infection, the VZV vaccine was found to be safe but less immunogenic than in healthy children, with seroconversion rates of approximately 60%. A recent phase II trial determined the VZV vaccine to be safe and immunogenic among HIV-infected children with a history of varicella, and no child had developed herpes zoster 2 years later. Fewer vaccine efficacy studies have been performed among HIV-infected adults: a boost in VZV-specific immunoglobulin G (IgG) was observed in HIV-infected patients on stable antiretroviral therapy for at least 3 months, with a nadir CD4 count greater than 400 cells/mL.
Among children who have received liver and intestine transplants and are on maximum dosages of 0.3 mg/kg prednisolone on alternate days and trough tacrolimus levels of less than 10 ng/mL for more than 1 month, the varicella vaccine was found to be safe, immunogenic, and protective. In the latter study, 16 children who were VZV IgG seronegative and aged between 13 and 76 months were immunized, and 87% and 86% of children developed humoral and cellular immunity, respectively. Five of the children developed a mild pain and erythema at the site of the injection, and another 4 developed fever and disseminated rash. Among 11 children with follow-up of 6 months or more after vaccination, there were 5 subsequent reported exposures to chickenpox (1 child with 2 exposures), none of which resulted in chickenpox.
A severe but nonfatal vaccine-associated disease has been reported in some children with an undiagnosed immunodeficiency. However, no fatalities have been observed in this group, in whom mortality associated with natural infection is known to occur. Hence, overall, it is believed that the potential benefits of vaccination against VZV in the immunocompromised host far outweigh the risks.
Other Viral Vaccines
Although HBV vaccination has been available for many years it has not been widely deployed in the setting of HBV infection in patients transplanted for chronic HBV. However, in the context of dialysis patients, HBV vaccination is recommended, although the immune response to vaccination can be suboptimal, especially if given intramuscularly. Recently, the use of the adjuvant 3- O -desacyl-4′-monophosphoryl lipid A (AS04) in conjunction with standard hepatitis B recombinant vaccine has led to enhanced immunogenicity (earlier and higher antibody titers) in dialysis patients.
At the time of writing, the prospects for viral vaccines against adenoviruses, BK virus, and HCV remain poor. It is therefore unlikely that prototype vaccines will be forthcoming for these infectious complications in transplant and oncology patients. Nevertheless, the experience and data generated from studies of HCMV and papillomavirus vaccines should provide important information concerning the likely immunogenicity of new vaccines in the context of dialysis and the immunocompromised state following transplantation.
Adoptive immunotherapy
Cytomegalovirus Immunotherapy
Antigen-specific T cells are essential for control of cytomegalovirus infection, and HCMV has evolved several mechanisms to overcome this component of the immune response. Immunotherapy offers an attractive way to improve immune reconstitution in transplanted patients, leading to control of viral replication without major side effects. As a consequence, this approach may reduce the use of antiviral chemotherapy and hence the adverse effects such as myelo- or nephrotoxicity and, furthermore, the evolutionary pressure on HCMV to develop drug resistance.
Various strategies to generate HCMV-specific T lymphocytes have been developed (these are reviewed in Ref. ). One of the most effective approaches for the ex vivo induction of HCMV-specific T cells is using CMV-infected fibroblasts, but the potential biologic risk that results from the use of live virus particles does not permit the use of infected antigen-presenting cells (APCs) for T cell stimulation under current good management practice (GMP) standards. Other approaches used to generate HCMV-specific T cells include HCMV peptide-pulsed dendritic cells (DCs), HCMV antigen–pulsed DCs, or using genetically modified APCs. All of these methods are effective in producing HCMV-specific T cells in high yield, but the process is expensive and time consuming to produce a product that can be deployed clinically.
Riddell and colleagues and Walter and colleagues were the first to show that adoptive immunotherapy for HCMV-specific CD8+ T cell clones into patients at risk of HCMV disease protected them from CMV-related complications. In these studies, 1×10 9 to 2×10 9 CMV-specific CD8+ T cells were administered and these cells persisted in patients’ blood for at least 8 weeks. HCMV prophylaxis was effective despite the progressive decline of these HCMV CD8 T cells in patients who did not develop a concomitant HCMV-specific CD4+ T Helper response. Although these results were groundbreaking, they suffered from the use of historical controls.
In an alternative study, patients lacking HCMV immunity were reconstituted by the adoptive transfer of HCMV-specific T cell lines which were generated by 4 repetitive weekly stimulations with HCMV lysate in vitro. These cells could be infused without any side effects, even in patients receiving their graft from a donor mismatched in 1 to 3 HLA antigens, implicating a potential use of this strategy even after haploidentical transplantation. This approach has advantages in the context of time compared with the generation of the CD8+ CMV–specific clones described earlier, and provides a more flexible application of the technique in patients with HCMV DNAemia at high risk for HCMV disease, without a loss of specificity and safety. These findings show that HCMV-specific adoptive immune transfer is a therapeutic option in patients who are DNAemic after stem cell transplantation and can be achieved by infusion of low numbers of CD4+ HCMV–specific T cell lines.
To facilitate the enrichment of virus-specific T cells, novel selection using the IFN-γ secretion assay conforming to current GMP regulations can be deployed. HCMV-Ag can also be used to elicit a combined HCMV-specific CD4+ and CD8+ T cell response. Stimulation of HCMV-specific CD4+ and CD8+ T cells was similarly effective when using HCMV antigen compared with the costimulation with HCMV antigen and HCMV peptides. As a result, an average of 1.3×10 8 CMV-specific stimulated, selected, and expanded T cells from 8/8 randomly selected HCMV-seropositive donors were generated in 10 days from a single 500-mL blood donation using only autologous cellular and humoral components compatible with current GMP regulations. The adoptively transferred T cells may then undergo further expansion in vivo if they are stimulated by HCMV-Ag–presenting cells providing an in vivo amplification step rather than relying on in vitro expansion.
Important questions remain with respect to the number of HCMV-specific T cells required for an effective adoptive transfer and their composition in respect to CD4+/CD8+ ratio and antigen specificity required for prevention or treatment of HCMV DNAemia after allogeneic stem cell transplantation. Further work is needed to define these parameters. However, improvements in immunologic assays have enabled direct quantification of HCMV-specific T cells. Analysis of phenotype and activity of antigen-specific T cells have contributed to a substantial improvement in understanding of the role and function of immune responses in vivo. Thus, phenotypic analysis with HLA-peptide multimers and functional assays can now be used in the setting of adoptive T cell therapy. Peptide-HLA multimers allow an easy visualization and isolation of antigen-specific cytotoxic T lymphocytes (CTLs). T cells that bind multimeric HLA complexes can be isolated to high purity using magnetic beads or fluorescent antibody cell separation (FACS) sorting. There are several reasons why the adoptive transfer of HCMV-specific CTLs freshly isolated from peripheral blood might be more efficient than the infusion of in vitro expanded and manipulated T cells. The process of in vitro expansion may increase the expression of the proapoptotic FAS molecule (CD95) and reduce telomere length of specific T cells, leading to a shorter survival of the adoptive transferred T cells similar to that observed in aging human populations. Various methods of cell generation result in different stages of T cell senescence. Freshly isolated and specifically selected T cells have greater expansion potential in vivo compared with repetitive in vitro stimulated T cells. However, specific stimulation ex vivo might reduce the risk for alloreactivity. In addition, the in vitro cell culture process increases not only the risk for contamination of the CTL preparation but also the costs for adoptive immunotherapy.
Although the availability of clinical-grade reagents for the selection of antigen-specific T cells has improved greatly in the last few years, not enough is known about the ideal composition of a cellular product that will be highly effective in use for adoptive transfer. The optimal targets for HCMV-specific T cell control have not been defined precisely despite evidence suggesting that the host makes a broad CD4 and CD8 response to most proteins in the HCMV proteome. However, there is still controversy about the benefit of transferring different T cell subsets for adoptive immunotherapy: namely, should only CD4+ T cells or CD8+ T cells be transferred, or a combination of both?
In view of the many different methods of generating T cells, future studies must address the following questions, preferably in placebo-controlled studies: (1) which are the best T cell subpopulations to be used for antiviral T cell therapy; (2) what differentiation/activation stage or stages are associated with efficient expansion and antiviral control; (3) what is the ideal range of antigen specificities of the T cells; and (4) what is the optimal cell dose required to control replication, and does it depend on the viral load and net state of immunosuppression of the patient?
EBV Immunotherapy
EBV is associated with posttransplant lymphoproliferative disease (PTLD), a common complication after hematopoietic stem cell and solid-organ transplantations. PTLD occurs in up to 15% of patients depending on the transplant type, age of the recipient, and the intensity of immunosuppression. With conventional treatments (reduction of immunosuppression, rituximab, radiotherapy, chemotherapy, surgery), relapses are common and the overall mortality remains high (around 50%).
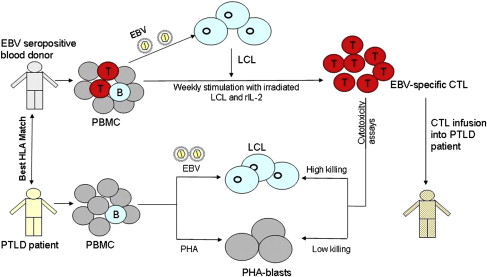
The search for an optimum treatment has led to recent trials of adoptive transfer of T cell immunity directed against EBV. These studies should be compared with those on HCMV discussed in the previous section. More than 90% of PTLD are EBV positive and most tumor cells express full latent cycle proteins (EB nuclear antigens [EBNAs] 1, 2, 3a, 3b, 3c, and leader protein [LP]; latent membrane proteins [LMPs] 1, 2), with a few cells expressing lytic cycle proteins. Some of these proteins, particularly the EBNA 3 family, are immunodominant targets for virus-specific CTL, thus making PTLD an ideal candidate for adoptive immunotherapy. EBV readily infects and transforms B lymphocytes in vitro giving rise to a continually growing lymphoblastoid cell line (LCL) in which each cell expresses all EBV latent antigens. Irradiated LCL cells can then be used as APCs to stimulate and expand autologous peripheral blood mononuclear cells (PBMCs) into an EBV-specific CTL line. Using this method, CTLs were grown ex vivo from HLA-matched hematopoietic stem cell donors and were used successfully to prevent and treat PTLD in respective stem cell recipients. However, this approach is not feasible in solid-organ transplantation because the organ donor is not generally available to provide blood, and the donor and the recipients are not closely HLA matched. Attempts have been made to generate autologous CTL from patients before transplantation and from patients with PTLD. However, the approach of generating autologous CTL from each transplant patient is prohibitively labor intensive and expensive for wide-scale use, and often there is not sufficient time to generate CTLs once PTLD has been diagnosed. An alternative strategy is to establish a bank of well-characterized EBV-specific CTL grown from EBV-seropositive healthy blood donors and provide these off-the-shelf CTLs to PTLD patients on a best HLA match basis ( Fig. 2 ). This third-party CTL strategy has several advantages: fully characterized CTLs are available for immediate use; 1 CTL line can be used for more than 1 PTLD patient, making it cost-effective; and these cells can be used to treat EBV-driven lymphomas in nontransplant immunosuppressed patients. A research team in Edinburgh University, Scotland, established a frozen bank of 100 EBV-specific CTL lines generated from Scottish blood donors selected to cover more than 99% of the HLA types of the UK population. In a pilot study, the first of its kind in the United Kingdom, 8 patients with progressive PTLD were infused with these allogeneic CTLs, selected on the basis of best HLA matches between the CTL donor and PTLD patient, with 3 patients achieving a complete remission. This CTL bank was then used in a multicenter phase II clinical trial to treat 33 PTLD patients (31 solid-organ and 2 stem cell transplant recipients in the United Kingdom, Sweden, France, and Australia) who had failed to respond to conventional treatments. At 6 months after treatment, 52% of patients had responded to the CTL infusions, with 14 achieving complete remission. A significantly better response was noted in patients receiving CTL with higher HLA matching ( P = .001) and with higher numbers of CD4+ T cells ( P = .001). No infusion-related toxicity or features of CTL-versus-host disease occurred in any of the infused patients and the third-party allogeneic CTL was considered a safe and effective form of treatment of PTLD.
Related posts:
 Purified Hematopoietic Stem Cell Transplantation: The Next Generation of Blood and Immune Replacement
Hematopoietic Stem Cell Transplantation: An Overview of Infection Risks and Epidemiology
Purified Hematopoietic Stem Cell Transplantation: The Next Generation of Blood and Immune Replacement
Hematopoietic Stem Cell Transplantation: An Overview of Infection Risks and Epidemiology
 Fungal Infections in Transplant and Oncology Patients
Fungal Infections in Transplant and Oncology Patients
 HLA-haploidentical Donor Transplantation in Severe Combined Immunodeficiency
Bone Marrow Transplantation Using HLA-Matched Unrelated Donors for Patients Suffering from Severe Combined Immunodeficiency
Infections in Pediatric Transplant Recipients: Not Just Small Adults
HLA-haploidentical Donor Transplantation in Severe Combined Immunodeficiency
Bone Marrow Transplantation Using HLA-Matched Unrelated Donors for Patients Suffering from Severe Combined Immunodeficiency
Infections in Pediatric Transplant Recipients: Not Just Small Adults
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


