The term lardaceous change has … come more into use chiefly through the instrumentality of the Vienna School…. The term, lardaceous changes … has but very little to do with these tumours, and rather refers to things, upon which the old writers … who were better connoisseurs in bacon than our friends in Vienna, would hardly have bestowed such a name…. The appearance of such organs … are said to look like bacon, bears … a much greater resemblance to wax, and I have therefore now for a long time … made use of the term waxy change…. These structures … by the simple action of iodine … assume just as blue a colour as vegetable starch….1
Schleiden, a German botanist, first used the term amyloid in 1838 to describe a normal constituent of plants.2, 3, 4 In 1858, Virchow, head of a pathology department in Berlin, gave a lecture entitled “Amyloid Degeneration” and described amyloid deposits that stained blue with iodine and sulfuric acid, similar to the chemical reaction markers of starch. Virchow concluded that the substance was composed of starch5 and used the word amyloid to describe it.6 During the lecture, Virchow also criticized his chief competitor in Vienna, Rokitansky, who believed that amyloid was a lardlike substance—perhaps because amyloid deposits were white and glistening—found in patients with syphilis, tuberculosis, or malaria.7 In 1859, Friedreich, Nikolau, and Kekule recognized that the waxy spleen described by Virchow did not contain any starchlike substances and that the deposits probably were derived from modified proteins.8 Kekule was famous in his own right at the time for describing the structure of benzene from his dream of a serpent biting its own tail. In addition, Friedreich gave the first description of a form of ataxia that now bears his name.9, 10
Budd analyzed the liver of a patient with amyloidosis and found that it was not lardaceous11; he also wrote original descriptions of rickets and scurvy.12 Probably the first reported case of primary amyloidosis (AL; described as “idiopathic” at the time) was identified by Wilks, who detailed a 52-year-old patient with lardaceous change unrelated to an obvious cause.13, 14, 15 Wilks was the first physician to use bromide in the treatment of epilepsy and wrote an original description of myasthenia gravis.
The amino acid composition of amyloid deposits was first described by Schmiedeberg in 1920.16 Amyloid proteins strongly resembled serum globulin and therefore were neither fat nor carbohydrate. The first use of Congo red as the specific stain for detection of amyloid was reported in 1922 by Bennhold.17 In 1927, Divry and Florkin18 reported green birefringence under polarized light when amyloid-laden material from the brain of a patient with Alzheimer disease was stained with Congo red. However, the association between the neurodegeneration of Alzheimer disease and amyloid was forgotten for nearly 50 years. Magnus-Levy19 postulated that Bence Jones proteins were a precursor of the amyloid substance and noted a relationship among amyloid deposits, Bence Jones proteins, and multiple myeloma.
The finding that amyloid proteins consisted of fibrils was credited to Cohen and Calkins in 1959.5 They determined that all forms of amyloid were nonbranching and fibrillar. The fibril length varied, but the width was 9.5 nm. Apitz20 claimed that amyloid in the tissues was analogous to the excretion of immunoglobulin (Ig) light-chain proteins by the kidneys; he coined the term paraprotein to describe monoclonal immunoglobulins. Isobe and Osserman21 reported in 1974 that Bence Jones proteins had a direct role in the pathogenesis of AL. Physiologically normal proteins primarily have an α-helix configuration. In 1968, Eanes and Glenner22 reported that they used x-ray diffraction to determine that amyloid proteins formed an alternate configuration of β-pleated sheets, similar to the configuration of silk proteins.23
As with silk, amyloid proteins are highly resistant to solvents, and this resistance is a feature of the purification process. Amyloid-laden tissue is homogenized repeatedly in saline and centrifuged. The supernatant, which contains soluble components, is discarded, and the residual pelletized material contains amyloid proteins. After the pellet is resuspended in distilled water, a relatively pure preparation of amyloid fibrils is obtained. Pras et al.24 first described the purification of amyloid in 1968. Levin et al.25 were the first to sequence amyloid protein, and they designated it as amyloid A. Benditt et al.26, 27 independently sequenced amyloid A at the same time. The first sequence of an immunoglobulin light-chain form of amyloid was reported in 1970 by Glenner et al.28 and was recognized as an N-terminal fragment of the immunoglobulin light chain.28, 29, 30, 31, 32
CLASSIFICATION
The diagnosis of amyloidosis requires biopsy tissue specimens with deposits that are positively stained by Congo red.33 With hematoxylin and eosin staining, amyloid deposits resemble hyalin. Deposits are always extracellular and appear amorphous. Apple-green birefringence is seen when Congo-red-stained material is viewed under polarized light.34 The Congo red stain can be technically difficult to use and can form precipitates, yielding false-positive results.35 Pathologists need to perform diagnostic assays regularly to be experts in the interpretation of stains. All forms of amyloid have a fibrillar appearance when viewed with electron microscopy, and the fibrils are rigid and nonbranching. However, not all fibrils identified with electron microscopy are amyloid proteins.36, 37, 38 Such findings are strongly suggestive of amyloidosis, but in the absence of a positive result on Congo red stain and apple-green birefringence, the diagnosis remains unconfirmed. Studies of recombinant-derived variable region fragments of immunoglobulins have shown a relationship between thermodynamic instability and fibrillogenic potential. Structural parameters and overall thermodynamic stability contribute to the fibril-forming propensity.39 Human monoclonal immunoglobulin light chains can be converted to amyloid fibrils in vitro by digestion with pepsin.30 Synthetically, amyloid fibrils can be produced by breaking the disulfide bonds of intact immunoglobulins. In the past, all forms of amyloid were thought to be derived from misfolded fragments of immunoglobulin light chains, but heavy-chain fragments have also been shown to produce amyloid (designated as AH).40
The classification scheme of amyloidosis has undergone revision as understanding of the pathophysiology of the disease has improved. In the 19th century, involvement of the liver, spleen, and kidneys was incorrectly thought to represent secondary amyloidosis (AA); amyloid that involved the heart, tongue, and peripheral nerves was classified as AL or idiopathic amyloidosis. Later, amyloidosis was classified by the site of first deposition and was referred to as pericollagenous or perireticular amyloidosis.41, 42 Familial amyloidosis (AF) was recognized generally by the presentation of progressive painful peripheral neuropathy with an autosomal dominant inheritance pattern.43, 44 Families with inherited renal amyloidosis were also described.45, 46 Mutations in transthyretin (TTR), apolipoprotein A1, fibrinogen A-α chain, lysozyme, leukocyte chemotactic factor 2 (LECT2),47 and apolipoprotein A2 are associated with hereditary amyloidosis. TTR amyloidosis is the most common form of AF, and it is associated with peripheral neuropathy.48
In the 19th century, when tuberculosis, leprosy, syphilis, and chronic infections were prevalent, patients with these infections often had concomitant AA. However, in the postantibiotic era, chronic inflammatory polyarthritis (e.g., ankylosing spondylitis, juvenile rheumatoid arthritis) is more commonly associated with AA. Crohn disease and chronic osteomyelitis have also been associated with a small number of AA patients in the Western hemisphere.49 Hereditary periodic fever syndromes, such as familial Mediterranean fever (FMF; the most common entity of this disorder), hyperimmunoglobulinemia-D syndrome, and tumor necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS), are characterized by recurrent fever and inflammation, and reactive amyloidosis is a complication of these hereditary periodic fevers. The causative genes of these syndromes have been identified. Anticytokine-based therapies are a new treatment option for these inflammatory conditions, and they potentially may prevent amyloidosis.50 Etanercept, adalimumab, and infliximab have all been reported to be effective for patients with AA.
One hundred years ago, the term primary amyloidosis referred to idiopathic amyloidosis and included all cases that were neither familial nor secondary; cases of unrecognized AF and AA for which a cause could not be established were mistakenly classified as AL. Currently, AL refers only to systemic or localized amyloidosis that is immunoglobulin light chain derived. A clonal plasma cell disorder exists for all patients with systemic AL, and the disease spectrum ranges from a barely detectable plasma cell clone to overt multiple myeloma.51 A classification of the various forms of amyloidosis is shown in Table 99.1.
TABLE 99.1 NOMENCLATURE OF AMYLOIDOSIS
Protein
Precursor
Clinical
AL or AH
Immunoglobulin (light or heavy chain)
Primary or localized amyloidosis; associated with myeloma or macroglobulinemia
AA
SAA
Secondary or familial Mediterranean fever; familial periodic fever syndromes associated with mutated tumor necrosis factor receptor
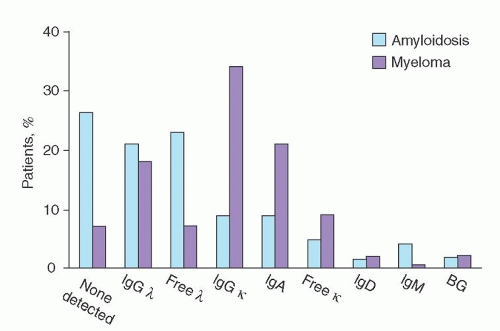
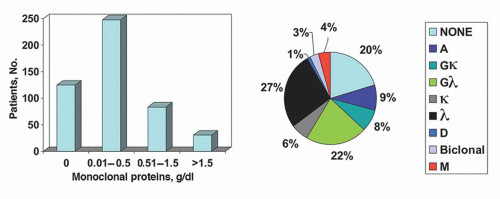
In AL, the tertiary structure and amino acid sequence of immunoglobulin light chains are abnormal.52, 53 For patients with AL, three fourths of immunoglobulin light chains are of the λ type. In contrast, for patients with multiple myeloma and monoclonal gammopathy of undetermined significance, two thirds of the light chains are of the κ type (Fig. 99.1).49 The implication is that λ immunoglobulin light chains have a greater tendency to form a β-pleated sheet. AL-associated immunoglobulins generally tend to have more β-pleated sheet proteins than α-helical proteins.
Certain immunoglobulins have “amyloidogenic” properties. No renal dysfunction is seen after mice are injected with purified immunoglobulin light chains derived from the urine of patients with multiple myeloma who do not have amyloidosis.54 However, deposits of human AL develop in the kidneys of mice after injection of light chains purified from the urine of patients with AL.
Amyloid-associated germline gene usage has been determined.55 The λ6 subgroup of immunoglobulin light chains is always associated with amyloid proteins, which suggests that unique amino acid sequences may result in formation of amyloidogenic proteins.56 The λ3 family appears most frequently in amyloidosis and polyclonal states. Two germline genes that belong to the λ3 and λ6 families encode 42% of λ variable regions. The gene segment 3r accounts for 22% of λ variable regions and is newly associated with the disease; gene segment 6a accounts for 20%. These gene segments have a strong association with amyloidosis; they are represented in less than 11% of polyclonal conditions (Table 99.2). Overuse of 3r and 6a likely accounts for the λ light-chain overrepresentation that is typical of amyloidosis. Germline gene usage may also influence the organ tropism of AL.57 Patients with clones derived from the 6a germline gene are more likely to present with dominant renal involvement. Those with clones from 1c, 2a2, and 3r are more likely to present with cardiac and multiorgan disease.58
The distinction between AL and amyloidosis associated with multiple myeloma is somewhat arbitrary (considerable overlap exists between them). However, a patient with amyloidosis and symptomatic multiple myeloma (e.g., widespread lytic bone disease, rib fractures, and lumbar spine compression fractures) is uncommon.59 Renal insufficiency in amyloidosis is almost never a consequence of the formation of light-chain cast nephropathy, as is the case in multiple myeloma. For patients with amyloidosis, renal failure is attributable to tubular atrophy, a consequence of long-term albuminuria.60, 61 Often, the distinction between multiple myeloma-associated amyloidosis and AL is made on the basis of the percentage of plasma cells in the bone marrow.62 For patients with AL, serial bone marrow biopsies performed over several years do not show a progressive increase in the plasma cell percentage. In amyloidosis, the process is clonal but typically is not proliferative, and the unrestrained growth associated with malignancy is absent. If it is not present at diagnosis, multiple myeloma does not develop subsequently in patients with AL who present with 10% to 30% bone marrow plasma cells; multiple myeloma develops in fewer than 0.5% of patients with amyloidosis.51 We have established an arbitrary threshold of 30% plasma cells to fulfill the criteria of multiple myeloma-associated amyloidosis if no other clinical features of multiple myeloma are present. For patients who have greater than 30% plasma cells in the bone marrow, the clinical course is dominated by AL and not by myeloma bone disease or myeloma-induced anemia.
FIGURE 99.1. Distribution of serum monoclonal protein in patients with amyloidosis (N = 270) or myeloma (N = 1,000). The κ-to-λ ratio is 1:3.6. BG, biclonal gammopathy. Ig, immunoglobulin. (From Gertz MA, Lacy MQ, Dispenzieri A, et al. Transplantation for amyloidosis. Curr Opin Oncol 2007;19:136-141 with permission from Lippincott Williams & Wilkins.)
aFor polyclonal conditions, λ1 usage is 25%, λ2 usage is 24%, and λ3 usage is 43%. Data from Perfetti V, Casarini S, Palladini G, et al. Analysis of Vλ-Jλ expression in plasma cells from primary (AL) amyloidosis and normal bone marrow identified 3r (λIII) as a new amyloid-associated germline gene segment. Blood 2002;100:948-953; Comenzo RL, Zhang Y, Martinez C, et al. The tropism of organ involvement in primary systemic amyloidosis: contributions of Ig V(L) germ line use and clonal plasma cell burden. Blood 2001;98:714-720; Abraham RS, Geyer SM, Price-Troska TL, et al. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain-associated amyloidosis (AL). Blood 2003;101:3801-3808. Epub 2002 Dec 19.
The incidence of AL is eight per million per year and has been stable for more than 50 years.63 Multiple myeloma is five times more prevalent than amyloidosis.64 Amyloidosis is uncommon; its incidence is similar to that of Hodgkin lymphoma (nodular sclerosing variant), chronic granulocytic leukemia (Philadelphiachromosome-positive), and polycythemia rubra vera.65, 66, 67 Bone marrow plasma cells from AL patients typically show chromosomal abnormalities.68 Trisomies of chromosomes 7, 9, 11, 15, and 18 are seen in 42%, 52%, 47%, 39%, and 33% of patients with amyloidosis, respectively. Fifty-four percent of male patients and 13% of female patients have trisomy X, and 72% have deletions in chromosome 18. The finding of aneuploidy in monoclonal plasma cells is indicative of their neoplastic nature, despite the fact that these plasma cells are nonproliferative and are present in small numbers (median, 5% plasma cells).69 An early pathogenetic event in the development of multiple myeloma is translocation at the immunoglobulin heavy-chain locus (band 14q32).70, 71 A total of 16 of 29 patients with AL (55%) showed translocations at the immunoglobulin heavy-chain locus. An additional 17% showed a pattern compatible with a possible IgH translocation.72 Overall, an IgH translocation was seen in 21 of 29 patients by using fluorescent in situ hybridization analysis. Of the 21 patients, 16 had t(11;14)(q13;q32) translocations. For 15 of the 16 patients, cyclin D1 overexpression accounted for 76% of all IgH translocations.
SYMPTOMS AND SIGNS
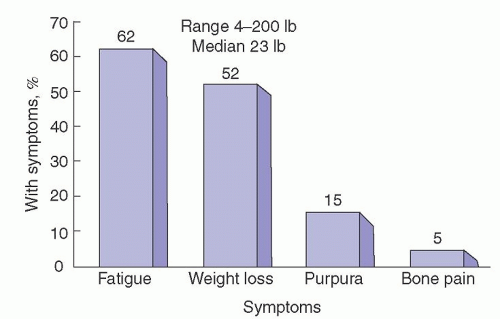
Weight loss, paresthesia, edema, dyspnea, and fatigue are the most common symptoms of AL (Fig. 99.2).73 These nonspecific complaints provide little help to a clinician evaluating patients. Patients with extreme weight loss frequently undergo evaluation for an occult malignancy. The fatigue, usually caused by early cardiac involvement with AL, generally is not associated with overt congestive heart failure and easily may be diagnosed incorrectly as stress-related or functional fatigue.74 We have seen many patients undergo coronary angiography because of fatigue and breathlessness, only to have the evaluation end when angiogram findings are normal.75
Light-headedness occurs frequently in amyloidosis, but this is a common and nonspecific complaint in the primary care setting. In AL, the cause of light-headedness is multifaceted.76 Patients with nephrotic syndrome have dizziness because of hypoalbuminemia and marked intravascular volume contraction, which leads to orthostatic hypotension.77 Patients with cardiac AL have a low end-diastolic volume (because of restriction to filling during diastole)78 and low cardiac output, but a normal ejection fraction is maintained until late in the course of the disease. The echocardiographic finding of a normal ejection fraction may be highly misleading and may hinder recognition of cardiac involvement in AL. Patients may have orthostatic hypotension as a consequence of autonomic neuropathy. Syncope is not unusual.79, 80
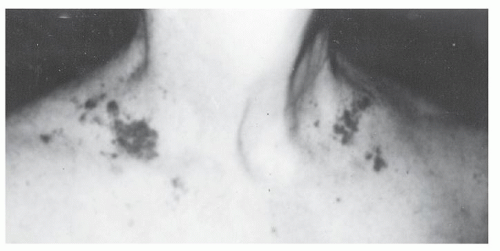
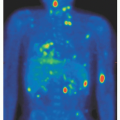
The physical findings of amyloidosis are specific and diagnostic; however, they are present in only 15% of patients and may easily be overlooked. Amyloid purpura is seen in only one of every six patients with AL (Fig. 99.3).81 Purpura may be periorbital but also may occur in the face, webbing of the neck, and upper chest. Purpura on the arms is not characteristic of AL, but petechial lesions on the eyelids should not be overlooked. The liver is palpable 5 cm below the right costal margin in only 10% of patients. Splenomegaly, if present, is usually of modest degree. Overall, any degree of hepatomegaly is present in one fifth of patients.82
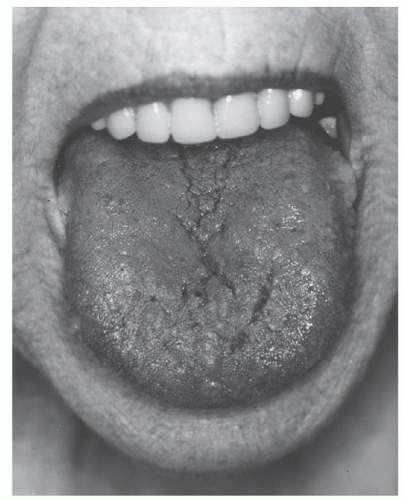
Macroglossia is the most specific finding of AL (Fig. 99.4). In our experience, enlargement of the tongue is never found in AF, AA, or senile systemic amyloidosis.83 Tongue enlargement is seen in 1 of 11 patients with AL and may be overlooked easily unless the physician knows to look for dental indentations on the underside of the tongue. However, presentation with fatigue, edema, breathlessness, or paresthesias would not immediately lead the physician to examine the patient’s tongue. Tongue enlargement is almost always accompanied by concomitant enlargement of the submandibular salivary glands,84 but salivary gland involvement should not be misinterpreted as submandibular lymphadenopathy. Major and minor salivary gland involvement may result in sicca syndrome.85
FIGURE 99.2. Prevalence of symptoms for patients with primary amyloidosis evaluated 1 month before or after diagnosis at Mayo Clinic, 1981-1992. (From Kyle RA, Gertz MA. Primary systemic amyloidosis. Clinical and laboratory features in 474 cases. Semin Hematol 1995;32:45-59. Used with permission.)
FIGURE 99.3. Classic truncal purpura in primary amyloidosis. (From Gertz MA, Lacy MQ, Dispenzieri A. Amyloidosis. Hematol Oncol Clin North Am 1999;13:1211-1233. Used with permission.)
Vascular involvement without visceral organ dysfunction produces occlusion and ischemic symptoms, such as jaw claudication, when the temporal arteries are involved86; calf and limb claudication may occur when the microvasculature that supplies the extremities is involved.87 Monoclonal protein in the serum may increase the sedimentation rate, and it is not unusual for amyloidosis and jaw claudication to be misdiagnosed as polymyalgia rheumatica.88 Amyloid deposits may be found if a temporal artery biopsy is performed, but Congo red staining typically is not performed on such specimens.87, 89 The shoulder-pad sign is a consequence of periarticular infiltration with amyloid and may produce pseudohypertrophy. Although the musculature of the shoulder and hip girdle is enlarged, patients present with diffuse muscular weakness90, 91 and may have muscular atrophy because of chronic vascular occlusion.
DIAGNOSIS OF AMYLOIDOSIS
When should a clinician initiate a diagnostic algorithm to confirm the presence of AL? The symptoms and physical findings of AL generally are nonspecific and unhelpful to the clinician. Nevertheless, eight critical clinical syndromes commonly associated with amyloidosis should trigger screening: (1) gastrointestinal tract symptoms of pseudo-obstruction or steatorrhea, (2) tongue enlargement, (3) carpal tunnel syndrome, (4) hepatomegaly, (5) peripheral neuropathy, (6) nephrotic-range proteinuria, (7) infiltrative cardiomyopathy with restrictive hemodynamics, and (8) atypical multiple myeloma. The diagnosis of AL must be considered when any one of these syndromes is seen (Table 99.3). AL also must be considered for any patient for whom the diagnosis of multiple myeloma is being considered and who has associated, unexplained weight loss or fatigue or a percentage of plasma cells in the bone marrow that does not meet the criteria for multiple myeloma.
FIGURE 99.4. Tongue enlargement in primary amyloidosis. (From Gertz MA, Lacy MQ, Dispenzieri A. Amyloidosis. Hematol Oncol Clin North Am 1999;13:1211-1233. Used with permission.)
TABLE 99.3 SYNDROMES IN PRIMARY AMYLOIDOSIS
Syndrome
Patients (%)
Nephrotic or nephrotic and renal failure
30
Hepatomegaly
24
Congestive heart failure
22
Carpal tunnel syndrome
21
Neuropathy
17
Orthostatic hypotension
12
A clonal population of plasma cells is observed in AL patients, even when the marrow percentage is less than 2%.92 The insoluble, fibrillar β-pleated sheets of amyloid proteins are derived from the monoclonal immunoglobulin light chains produced by a clonal population of plasma cells. For any patient with a compatible clinical syndrome that includes monoclonal gammopathy, tests to confirm the diagnosis of AL should be pursued aggressively. Important screening tests for a patient with cardiomyopathy, neuropathy, hepatomegaly, or proteinuria are immunofixation of the serum and of the urine93 and immunoglobulin free light-chain measurement.94 The urine must be evaluated because serum assays may fail to detect monoclonal light chains (Fig. 99.5). Serum electrophoresis tests are inadequate because the light chains in AL are frequently present only in trace amounts and they do not produce a spike on an electrophoretic pattern. Monoclonal protein in the urine frequently is difficult to detect because most patients have proteinuria to the extent that small amounts of light chains are obscured (Fig. 99.6). If amyloidosis is being considered in the differential diagnosis, immunofixation of serum or urine (or both) is essential. The immunoglobulin free light-chain assay is tenfold more sensitive than serum immunofixation for the detection of serum light chains.95 If all three tests are negative, systemic AL is not present.
For patients with a suggestive clinical syndrome, immunofixation is a good noninvasive screening test. For patients with light-chain levels in the serum or urine that are below the threshold of detection by immunofixation,96 the bone marrow often shows a clonal population of plasma cells when examined by flow cytometry, immunofluorescence, or immunohistochemistry.97, 98 Such patients frequently have abnormal κ-to-λ ratios of free light chains. In patients with confirmed AL who did not have detectable levels of monoclonal protein in the serum or urine, 94% had a clonal population of plasma cells in the bone marrow when a slide-based immunofluorescence test was used to detect expression of clonal cytoplasmic immunoglobulin. If the patient does not have monoclonal protein in the serum or urine, has a normal free light-chain ratio, and has no detection of clonal population of plasma cells in the bone marrow, the diagnosis of systemic AL is very unlikely. If amyloidosis is present, it likely is not immunoglobulin light-chain derived, and further evaluation for familial, secondary, or localized amyloidosis should be performed.
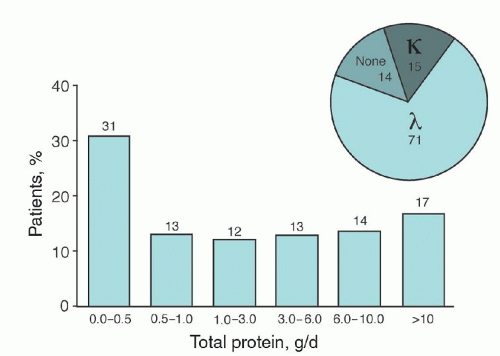
FIGURE 99.5. Serum monoclonal protein levels from patients with primary amyloidosis (N =488). Inset, immunofixation results.
Our ability to recognize AL has been improved by the nephelometric assay for free light chains in the serum; the assay detects free immunoglobulin light chains that are not associated with an intact immunoglobulin molecule.99, 100 For 100 consecutive AL patients tested with the nephelometric assay, the assay sensitivity was 90% for patients with confirmed AL-κ, and similar sensitivity was noted for patients with confirmed AL-λ. For patients with detectable urinary light chains (but no detectable monoclonal protein in the serum with immunofixation assays), the nephelometric light-chain assay detected free light chains in the serum of 85% of patients with AL-κ and 80% of patients with AL-λ. For patients with AL who had no monoclonal protein detected in the serum or urine with immunofixation assays, the nephelometric light-chain assay identified free κ light chains for 86% of patients with AL-κ and free λ light chains for 30% of patients with AL-λ. The free light-chain serum assay is convenient and adds an important diagnostic tool for classifying and monitoring AL patients.
All forms of amyloid deposits contain the amyloid P component, a pentagonal glycoprotein that may represent as much as 10% of the amyloid fibril by weight. It is structurally similar to C-reactive protein and has been identified in all vertebrates. However, it does not function as an acute-phase reactant in humans because the concentration in plasma is relatively stable.101 Amyloid P component maintains a dynamic circulating equilibrium between the serum and amyloid deposits. Because serum amyloid P (SAP) exchanges readily with the amyloid P component of amyloid fibrils, binding and release of SAP from amyloid deposits may be observed with an 123I-labeled SAP scan.102, 103 In addition, the clearance rate of radiolabeled P components (125I-labeled SAP) from the plasma may be used to assess the total-body burden of amyloid and to evaluate the effect of therapy.104 Patients with large burdens of amyloid have rapid clearance, which is associated with shorter survival, but those with trace amounts of systemic amyloid deposits have plasma clearance rates similar to those of healthy individuals. Healthy adults have 50 to 100 mg of amyloid P component in the extravascular and intravascular compartments, whereas patients with amyloidosis may have up to 20,000 mg. Antibodies to P component have been used as therapy against amyloidosis.105
FIGURE 99.6. Total protein excreted in the urine of patients with primary amyloidosis (24-hour period). Inset, identification of immunoglobulin light chains, %. (From Gertz MA, Lacy MQ, Dispenzieri A. Amyloidosis. In: Mehta J, Singhal S, eds. Myeloma. London: Martin Dunitz, 2002:445-463. Used with permission.)
The P component scan may be performed serially to determine whether patients have increasing rates of amyloid deposition. However, the technique does not determine whether the amyloidosis is AL, AF, AA, or localized. Cardiac amyloid deposits are not detected through the use of SAP scans,106 but uptake of 123I-labeled amyloid P component is seen in the spleen, liver, and kidneys in 87%, 60%, and 25% of patients, respectively. Imaging does not consistently show deposits in the carpal tunnel, kidneys, and gastrointestinal tract. The correlation is poor between imaging and the extent of organ dysfunction assessed clinically.
The diagnostic sensitivity of SAP scintigraphy for systemic AA, AL, and amyloid TTR origin amyloidosis is 90%, 90%, and 48%, respectively; specificity is 93%. Splenic amyloid is observed in 80% of AL patients, even though it rarely is detected clinically. Bone marrow uptake is specific for AL in 21% of patients. More widespread organ involvement is identified with a scan than with a clinical examination.107 Amyloid is distributed heterogeneously within individual organs. Presently iodine-labeled SAP scanning was not available in the United States.
The diagnosis of amyloidosis must always be confirmed by tissue biopsy.108 For patients who have neuropathy, nephrotic syndrome, cardiac failure, or hepatomegaly, the diagnosis could be established by direct biopsy of the involved organs,108 but an invasive visceral biopsy usually is not required to confirm AL. Because amyloid deposits typically are widespread at diagnosis and have involved the vasculature extensively,109 biopsy procedures that are less invasive, lower risk, and lower cost may be used to establish the diagnosis.
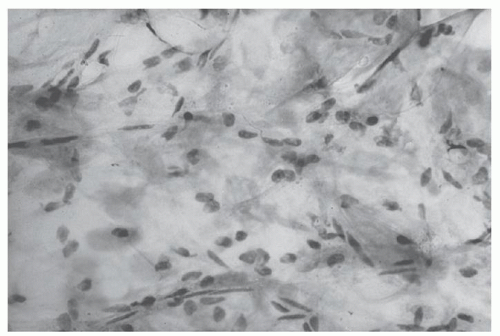
For a patient with a syndrome consistent with AL and confirmed presence of monoclonal protein, we begin the diagnostic evaluation with a subcutaneous fat aspirate and bone marrow biopsy.110 The subcutaneous fat aspirate is a 75%-sensitive, riskfree procedure that is performed by registered nurses.111 Results are available within 24 hours (Fig. 99.7). Amyloid deposits are seen in half of the bone marrow biopsy specimens of patients with AL and generally are identified in blood vessels.112 To exclude the diagnosis of multiple myeloma, a bone marrow biopsy is justified for any patient who has monoclonal immunoglobulin light chains. Knowledge of the percentage of bone marrow plasma cells is important prognostically.113Table 99.4 shows the findings of bone marrow biopsies and fat aspirations of patients with AL. With Congo red studies of fat and bone marrow, the diagnosis is established in 87% of patients. For the other 13%, the diagnosis may be established by a biopsy of the affected organ.
Less invasive biopsy procedures may be used to establish a diagnosis of AL. Xerostomia in amyloidosis is attributable to salivary gland infiltration, and lip biopsy findings have high sensitivity (up to 87%).114, 115 Subcutaneous blood vessels may be accessed through a skin biopsy and may show AL deposits.116, 117 Amyloid deposits regularly were confirmed by rectal biopsy in the 1970s and 1980s.118 Rectal biopsy is an outpatient procedure, although occasionally it results in bleeding. Endoscopic biopsy specimens frequently are inadequate because submucosa is essential for specimen adequacy.
FIGURE 99.7. Subcutaneous fat aspirate showing amyloid deposits (congo red, original magnification ×100). (Courtesy of Paul J. Kurtin, MD, Mayo Clinic, Rochester, Minnesota. Used with permission.)
All patients who have nephrotic syndrome should undergo screening with immunofixation assays of serum and urine and free light-chain assays. Among 110 patients with AL, the sensitivity of the free light-chain assay was 92%, but in combination with serum and urine immunofixation assay findings, the sensitivity increased to 99.1%.119 If either assay finding is positive, a fat aspiration is performed, which eliminates the need for a diagnostic renal biopsy for 70% of patients. The net result is reduced expense and reduced risk of bleeding and hospitalization.
Congo red may precipitate in tissue, and the resultant overstaining, particularly in subcutaneous fat, may produce a false-positive result.120 Skin and subcutaneous fat have a high content of elastin and collagen; both bind Congo red and may also be interpreted as a false-positive result.121 Rectal biopsy specimens containing amyloid may be misinterpreted as collagenous colitis if stained only with hematoxylin and eosin.122 To recognize amyloid in the myocardium, our cardiac pathologists prefer to use sulfated Alcian blue stain.123 The Peripheral Nerve Laboratory at Mayo Clinic stains sural nerve biopsy specimens with crystal violet during screening and confirms the diagnosis subsequently with Congo red.124
DIFFERENTIATING AMYLOIDOSIS FROM OTHER FORMS OF AMYLOIDOSIS
It is important that the diagnosis of amyloidosis be confirmed as AL, AF, or AA because therapy for the three syndromes differs.125, 126 Localized amyloidosis,127 AF,128 AA,129 and senile systemic forms of amyloidosis130 are not associated with a plasma cell dyscrasia. Except for the rare case of a patient who has one of these forms of amyloidosis and an incidental monoclonal gammopathy of undetermined significance, only AL patients have monoclonal light chains in the serum and urine or clonal plasma cells in the marrow. Patients with localized amyloidosis may present with hematuria,131 respiratory difficulties,132 and visual disturbances133; such symptoms may easily be confused with those of systemic amyloidosis. The localized amyloidosis syndrome, which usually involves the skin, tracheobronchial tree, or urogenital tract, never becomes systemic. The fibrils of localized amyloidosis may be immunoglobulin light-chain derived,134 but clonal plasma cells are not seen in the bone marrow.
The site of amyloid deposition provides an important clue for recognizing localized amyloidosis. The most frequent sites of localized amyloid deposits are the respiratory tract, genitourinary tract, and skin.126, 135 Pulmonary amyloid may be subdivided into nodular, tracheobronchial, or diffuse interstitial deposits. Only the last type of deposit is a manifestation of systemic AL.136 The diagnosis of tracheobronchial amyloidosis is made by bronchoscopy during evaluation of a patient with obstruction, cough, dyspnea, wheezing, or hemoptysis. The usual treatment is resection of the tissue with an yttrium-aluminum-garnet laser.137 Tracheobronchial amyloid deposits are derived from immunoglobulin light chains.138 Nodular amyloidosis presents as a solitary pulmonary nodule or multiple nodules, and the nodules do not indicate systemic AL.139 Nodules are not calcified, and they often require resection to exclude a diagnosis of malignancy. The AL diagnosis is established during a thoracotomy or a video-assisted thoracoscopic surgical procedure. Amyloid can involve the vocal cords and false vocal cords and cause traction on the structures, leading to hoarseness. This form of laryngeal amyloidosis is always localized.140
TABLE 99.4 FINDINGS OF NONINVASIVE BIOPSIES IN PRIMARY AMYLOIDOSIS
Biopsy Finding
Fat
Marrow
Patients (%)
+
+
55
+
–
22
–
+
10
–
–
13
+, tissue positive for amyloid deposits; -, tissue negative for amyloid deposits. Modified from Gertz MA, Lacy MQ, Lust JA, et al. Prospective randomized trial of melphalan and prednisone versus vincristine, carmustine, melphalan, cyclophosphamide, and prednisone in the treatment of primary systemic amyloidosis. J Clin Oncol 1999;17:262-267. Used with permission.
Obstructive ureterovesicular amyloidosis is always localized. Patients present with hematuria and have a prebiopsy diagnosis of cancer. Amyloid deposits are found when cystoscopic biopsies are performed. Eighty-five percent of patients with this type of amyloidosis present with hematuria. Patients may undergo partial cystectomy, fulguration, or transurethral resection.141 Dimethyl sulfoxide (DMSO) instillation in the bladder has been reported to reduce these deposits.142 Colchicine has also been reported to be beneficial. Amyloid involving the renal pelvis or ureter is a localized amyloid syndrome.143, 144 These patients present with colic because of obstruction or hematuria, and deposits are found during surgery. Nephrectomy is commonly performed because the ureteral mass preoperatively is thought to represent a transitional cell malignancy, but the recognition of amyloidosis avoids nephrectomy. Patients may present with dysuria and hematuria when amyloidosis involves the urethra.145 The preoperative diagnosis is usually a urethral malignancy. Resection is the treatment of choice.
Three forms of cutaneous amyloidosis are recognized. The lichen and macular forms are localized,146 innocuous conditions that usually are associated with a history of local skin trauma or inflammation. Nodular amyloidosis is associated with AL,147 and evidence of nodular deposits may be an important clinical clue to an underlying life-threatening process. Degraded keratin molecules are the source of macular and papular amyloid deposits.148 Dermabrasion and other forms of local therapy adequately control cutaneous amyloidosis.
Carpal tunnel amyloidosis may be observed in systemic AL and AF, but it may also be localized.149, 150 For patients who present with carpal tunnel syndrome as the only manifestation of amyloidosis, the median survival is 12 years. Virtually all patients with carpal tunnel amyloidosis have localized disease, and one study reported that only 2 of 124 patients with localized carpal tunnel amyloidosis had development of AL.151 TTR is often found in localized carpal tunnel amyloid deposits.
Localized amyloidosis is seen in the conjunctiva and orbits. The best treatment is surgical excision. Amyloid has also been localized to breasts,152 mesenteric lymph nodes, colonic polyps, thyroid,153 retroperitoneum, and ovaries. Localized deposits of amyloid commonly are observed in trace amounts within the cartilage on the hip surface after a total hip arthroplasty.154 Similarly, localized deposits are found in resected knee arthroplasty specimens. These deposits are not associated with systemic disease.155
AA is a consequence of poorly controlled, long-term systemic inflammation. From a simple syndrome standpoint, its presentation is similar to that of AL. AA is more common than AL in underdeveloped countries because of the persistence of tuberculosis,61 syphilis, malaria, and leprosy, but in the United States, AA is not easily distinguished from AL. AA is not associated with a monoclonal protein or clonal marrow plasma cells. Patients with AA most commonly present with nephrotic-range proteinuria.156 At Mayo Clinic, AA accounts for only 2% of all patients with amyloidosis.157
The underlying cause of AA most commonly includes ankylosing spondylitis,158 juvenile rheumatoid arthritis,159 psoriatic arthritis,160 and rheumatoid arthritis161; for most patients with AA, the cause is clear because the arthritis is disabling and develops a median of 15 years before the development of AA.162 Of patients with rheumatoid arthritis, AA develops in 3.1%.163 With the introduction of anti-TNF therapies for inflammatory arthropathy, the prevalence of AA in industrialized nations continues to decrease as these disorders are better controlled. AA is also seen in patients with Crohn disease,164 bronchiectasis165 (including long-term survivors of cystic fibrosis), and chronic osteomyelitis.166 These are conditions in which the infected tissue is not amenable to surgical excision and antibiotic therapy results in poor control. For most patients with AA and Crohn disease, the inflammatory bowel disorder has been present for decades. The first clinical manifestation is proteinuria. No monoclonal protein is detectable. AA has been described in individuals who subcutaneously inject contaminated illegal substances (i.e., “skin poppers”).167, 168 Skin abscesses develop at the site of injection and cause the inflammation that is necessary for the development of AA. Patients with Hodgkin lymphoma169, 170 or hypernephroma171, 172 and paraplegic patients with chronic infected decubitus ulcers or chronic urinary tract infection173, 174 have been reported to have AA. Amyloid deposits are identified during autopsy for half of all patients who sustained a spinal cord injury 10 years or more before death.175 AA from Castleman disease has been recognized. Surgical excision leads to remission.176, 177 AA has also been reported for patients with hyperimmunoglobulin D syndrome and periodic fever syndromes, so-called TRAPS.178
AF is more common than AA in the United States. Presentation of AF is clinically indistinguishable from AL; patients may have cardiomyopathy,179, 180 neuropathy,181 and nephrotic syndrome.182 The most common form of AF is attributable to TTR mutations. More than 100 different mutations in the TTR gene have been involved in amyloid neuropathy.183 Nearly half of the patients with AF seen at Mayo Clinic do not have a family history of the disease, but absence of a family history is a poor screening method to exclude AF.184 The most common TTR mutation seen at Mayo Clinic is Thr60Ala, caused by a nucleotide substitution in TTR DNA of 238A>G. Amyloid neuropathy without monoclonal protein or clonal plasma cell disorder should raise clinical suspicion of AF. Cardiac amyloid deposition may occur with mutated and wild-type TTR genes.185 Cardiac amyloid deposits186 that consist of normal TTR proteins occur in 8% to 25% of people older than 80 years. In this form of senile systemic amyloidosis, only cardiac symptoms are seen.
AF involving only the heart (without peripheral neuropathy) was first described in a Danish kindred.187 Since then, AF cardiomyopathy has been described throughout the world. The US patients in AF pedigrees usually have symptoms after age 60 years. Patients with cardiomyopathy from AF present with heart failure or arrhythmias, and the clinical picture is virtually indistinguishable from that of senile cardiac amyloidosis or AL. Twenty-one percent of patients older than 90 years (the general patient population) have senile systemic amyloidosis from deposition of wild-type TTR.188 The Val122Ile mutation of TTR is a major cause of inherited cardiac amyloidosis for African Americans. The first report of the mutation described an African American man aged 68 years.189 The TTR Val122Ile allele is carried by 3.9% of African Americans, which translates to 1.3 million people in the United States.190 Therefore, a finding of cardiac amyloidosis without monoclonal gammopathy could represent AF-based cardiomyopathy (even without a family history), and clinicians should not assume that the patient is affected by AL.191
Mutations in the fibrinogen,192 lysozyme,193 or apolipoprotein A1 and A2194, 195, 196 and LECT2 proteins may produce inherited forms of renal amyloidosis. The amyloidosis associated with these mutations has a much better prognosis than that of renal AL. We have regularly seen patients who have proteinuria for more than a decade without renal failure. Because these patients present with proteinuria and the renal biopsy specimen shows amyloid deposits, their condition is easily confused with AL. The absence of a monoclonal immunoglobulin disorder or the absence of free light chains in the serum is an important distinguishing feature. However, only immunohistochemical staining or sequencing of the amyloid can differentiate these entities definitively. Of 285 renal amyloid samples, 31 were unclassified and subsequently 7 were found to be LECT2 related by tandem mass spectrometry.197 Isolation of genomic DNA and polymerase chain reaction amplification of LECT2-encoding exons showed no mutations. However, all were homozygous for the G allele encoding valine at position 40 in the mature protein. LECT2-associated renal amyloidosis represents a unique form of renal amyloidosis, especially in Mexican Americans, and has only recently been recognized.
The treatment of TTR-derived AF includes liver transplantation, which makes the distinction between AF and AL critical for treatment decisions.198 TTR is produced in the choroid plexus and the liver. Regression of amyloid deposits has been reported199 when liver transplantation is performed before the development of disabling peripheral neuropathy, autonomic neuropathy, or advanced cardiomyopathy. Patients with the Val30Met mutation in the TTR gene appear to have the best outcome after liver transplantation.200 Progressive cardiac amyloidosis has been reported after liver transplantation for patients with other TTR mutations.201 After mutant TTR is deposited in the myocardium, it may serve as a nidus for further deposition of native TTR produced by the transplanted liver. Autopsy findings show cardiac amyloid fibrils with mutant and wild-type TTR.202, 203 AF may be initially diagnosed as AL unless the patient has a clear family history of AF. When the referral diagnosis was AL, 34 of 350 patients (9.7%) were reported to have AF.204 The most common mutation in AF was identified in the gene encoding the fibrinogen A-α chain, and the second-most common mutation occurred in the gene encoding TTR. A low-grade monoclonal gammopathy was detectable in eight patients. All patients who have clinical syndromes consistent with AF should be screened for a genetic cause when AL cannot be confirmed unequivocally.204 Of 178 consecutive patients referred for amyloidosis, 54 underwent screening with polymerase chain reaction assays to detect AF variants.205 Three patients had monoclonal gammopathy and a hereditary variant; this finding justified routine screening of patients with apparent AL for hereditary variants and showed the necessity of immunohistochemical or mass spectroscopic confirmation of the type of amyloid protein subunit.
Screens for mutant forms of TTR may be performed using immunoaffinity chromatography and immunoprecipitation; both are convenient methods for assessing circulating TTR in serum.206 In addition, TTR variants may be examined using immunoprecipitated serum proteins and matrix-assisted laser desorption ionization mass spectrometry.207
The gold standard for protein identification is mass spectroscopic analysis of the tissue and direct sequencing to identify the amyloid protein, and this evaluation is now standard for all pathologic specimens that test Congo red positive seen at Mayo.208, 209 The procedure can be performed on subcutaneous fat. The distinction between immunoglobulin and nonimmunoglobulin amyloidosis is important because nonimmunoglobulin amyloidosis does not benefit from chemotherapy.
In summary, any patient with amyloidosis who does not have a detectable monoclonal light chain in the serum or urine and does not have a clonal population of plasma cells in the bone marrow should be considered for AA, AF, or localized amyloidosis. Laser capture microdissection mass spectroscopic analysis is the most direct method to identify the protein subunit comprising the amyloid and is recommended for all positive tissue biopsies.
PRESENTATION AND CLINICAL FEATURES
We reviewed records of all patients who received a diagnosis of AL over a 10-year period at Mayo Clinic. All patients received the diagnosis within 30 days of presentation at our institution. All had a clonal plasma cell disorder and histologic proof of amyloid deposits. Patients with overt multiple myeloma were excluded. The male preponderance of AL (67.3%) has been constant at our institution for 40 years, and we believe this finding reflects a true higher prevalence of the disorder in men. Multiple myeloma also is more common in men, but the male-to-female ratio is 52:48. Although we have seen patients with amyloidosis who were as young as 27 years, the median age of patients with amyloidosis was 67 years (range, 39 to 89 years). The median age of patients who present with AL in Olmsted County, Minnesota, is 73 years63; this age difference suggests that a referral bias to amyloidosis treatment centers may exist.
Echocardiographic examinations have dramatically increased the ability to recognize cardiac amyloid deposits.210, 211 Cardiac amyloidosis was seen in 37.4% of patients with AL at Mayo Clinic. Congestive heart failure was present in only half of these patients; for the other half, the presenting symptoms of cardiac amyloidosis were fatigue and dyspnea. These patients had poor cardiac filling and low cardiac output, but the ejection fraction was preserved. Syncope and arrhythmias also occurred.
Renal amyloid deposits were seen in 30% of patients with AL; kidneys were the second-most common organ affected by AL.212 Nephrotic-range proteinuria was observed for 95% of patients with renal AL; the other 5% had amyloid deposits in the interstitium and mesangium of the kidney but not the glomeruli, so proteinuria was not present. Sensorimotor symmetrical ascending peripheral neuropathy was present in 15.3% of AL patients.24 Palpable hepatomegaly was observed in 17.7% of AL patients, but a dominant hepatic syndrome was seen in only 4.6%. Gastrointestinal tract amyloidosis occurred for 7.1% of patients with AL, and they presented with intestinal bleeding, pseudo-obstruction, or diarrhea. The other 7.8% of patients had amyloid deposits that were identified in only the tongue, lung, joints, or soft tissues.
At Mayo Clinic, 29.9% of AL patients had two or more organs affected, 6% had three affected organs, and 0.5% had four or more affected organs.213 When multiple organs were involved, the liver was most likely to be affected. Clinically significant bleeding (excluding purpura) occurred for only 2.3% of our patients.214, 215 Congestive heart failure was observed for 22% of patients, and carpal tunnel syndrome was observed for 21% of patients.212 Edema was noted for 44.8% and fatigue for 46.4%. Lower-extremity paresthesias, nephrotic-range proteinuria, orthostatic hypotension, and weight loss were observed for 34.9%, 21.0%, 12.5%, and 51.7% of patients, respectively. The median survival of our entire cohort was 12 months, and the 2- and 5-year survival rates were 33.6% and 14.9%, respectively.
Amyloidosis is rarely associated with anemia.216 The hemoglobin level was greater than 100 g/L for 90% of patients and greater than 120 g/L for 64.4%. Only 1.5% had hemoglobin levels less than 90 g/L, and the cause was usually active gastrointestinal tract bleeding or renal failure. The median platelet count of patients with AL was 257 × 109/L (range, 46 × 109/L to 809 × 109/L). A platelet count greater than 500 × 109/L was seen for 5.5% of patients. The most common cause of thrombocytosis in AL is hepatic involvement with associated hyposplenism.217, 218 A serum creatinine value of 2 mg/dl or greater was present for 13.6% of patients, and 6.7% had alkaline phosphatase levels greater than twice the maximum normal value. The number of patients with elevated alkaline phosphatase levels was close to the number of patients with clinical hepatic amyloid involvement.
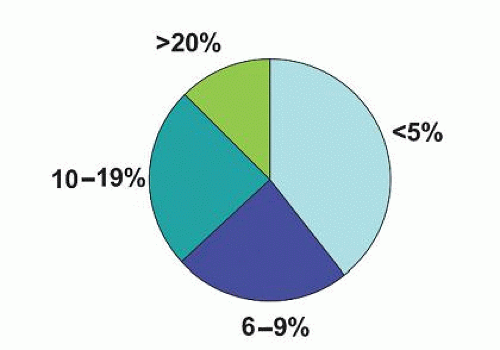
The median bone marrow plasma cell percentage of patients with AL is 7% (range, 1% to 30%; Fig. 99.8).212 In 11.3% of patients with AL, greater than 20% of plasma cells were found in the bone marrow, but no other clinical evidence of multiple myeloma was observed. Proteinuria is exceedingly common in AL, and the median urine protein loss is 790 mg per 24 hours (Fig. 99.6).219 Monoclonal λ and κ light chains (in the serum and urine) were found in 70.1% and 18.9% of patients, respectively, and 11% had no detectable light chains. For patients with intact monoclonal γ globulins, the serum heavy chain was IgG, IgA, and IgM for 58%, 10%, and 8.2% of patients, respectively. The presence of an IgM monoclonal protein in 8.2% is important because we have seen many referred patients with a diagnosis of Waldenström macroglobulinemia, for whom the amyloid syndrome was overlooked.220 Amyloid arthropathy occasionally occurs in amyloidosis. Synovial membrane biopsy may establish the diagnosis, and chemotherapy may effectively alleviate the joint manifestations.221
Heart
The heart is the organ that determines the patient outcome, and it is the organ most frequently involved in AL.222, 223, 224 Amyloid is deposited extracellularly and results in a noncompliant and thickened left ventricle (Fig. 99.9).225, 226 The clinical presentation is infiltrative cardiomyopathy with restricted diastolic ventricular filling. Many patients with cardiac AL present only with fatigue and unexplained weight loss. Early cardiac AL produces diastolic dysfunction without systolic dysfunction,227, 228 and chest radiographs do not show evidence of pulmonary vascular congestion or cardiomegaly. Moreover, the ejection fraction is preserved because amyloid deposition is an infiltrative process with diastolic dysfunction only.229 Indeed, because of a low stroke volume, a hyperdynamic myocardium may develop with an elevated ejection fraction. This constellation of findings is frequently misinterpreted, and the presence of amyloid may be overlooked completely. An echocardiographic examination usually shows that contractility is normal, but because of poor diastolic filling, the end-diastolic volume is reduced and cardiac output is low. The electrocardiogram shows low voltage, but this also may be easily overlooked.230 The pseudoinfarction pattern of amyloidosis, with nearly two thirds of patients showing loss of anterior forces in V1 through V3,230 may be misinterpreted as ischemic heart disease. Electrocardiographic findings are clinically suspicious of silent ischemic disease, and patients invariably undergo coronary arteriography. Coronary angiogram findings are generally normal.231 A characteristic echocardiogram shows wall and valve thickening, diastolic dysfunction, and a sparkling myocardium (Fig. 99.10). The median septal thickness for patients with AL was 14 mm.210 The septal wall thickness was less than 15 mm for 52.9% and greater than or equal to 15 mm for 47.1%.210 Wall thickening frequently is misinterpreted as concentric left ventricular hypertrophy or asymmetrical septal hypertrophy, rather than infiltrative cardiomyopathy.232, 233, 234 When ventricular hypertrophy is diagnosed from the thickened walls, the electrocardiogram typically shows low voltage and does not reflect hypertrophy.235 Screening tests for monoclonal protein in the serum or urine should be performed for any patient with diastolic heart failure, restrictive hemodynamics, or thickened walls.
FIGURE 99.8. Distribution of bone marrow plasma cells of patients with primary amyloidosis (N= 486).
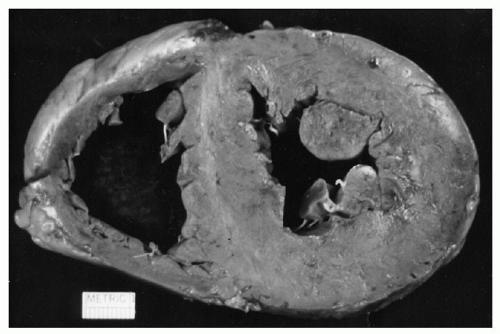
FIGURE 99.9. Myocardial wall diffusely thickened with amyloid. The whitish deposits represent the amyloid and are the lardaceous changes first recognized by Rokitansky. (From Gertz MA, Kyle RA. Amyloidosis [AL]. In: Wiernik PH, Goldman JM, Dutcher JP, et al., eds. Neoplastic diseases of the blood, 4th ed. New York: Cambridge University Press, 2003. Used with permission.)
Patients with cardiac amyloidosis have restriction to blood inflow that is characteristic of the disease. Doppler studies are best for accurate assessment of myocardial function for AL patients. The Doppler filling patterns accurately indicate the extent of amyloid infiltration.236 For patients with advanced cardiac amyloidosis, a short deceleration time is consistent with restrictive physiologic characteristics, but in the early stages of cardiac amyloidosis, abnormal relaxation is observed.237 Decreased fractional shortening and increased ventricular wall thickness are the best predictor of outcome in AL. Patients with a wall thickness greater than 15 mm and a fractional shortening less than 20% have a median survival of 4 months. An important measurement is the deceleration time; a short deceleration time is indicative of restrictive physiologic characterisitics, and a deceleration time shorter than 150 milliseconds is associated with worse outcomes than that of a deceleration time longer than 150 milliseconds.
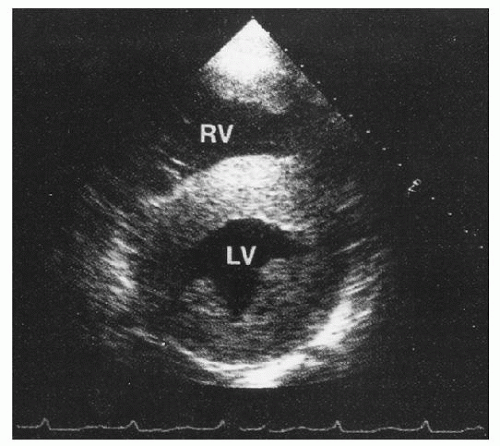
FIGURE 99.10. Echocardiographic image of concentric thickening of the wall of the left ventricle (LV) due to amyloid. RV, right ventricle. (From Gertz MA, Kyle RA. Amyloidosis [AL]. In: Wiernik PH, Goldman JM, Dutcher JP, et al., eds. Neoplastic diseases of the blood, 4th ed. New York: Cambridge University Press, 2003. Used with permission.)
The most common echocardiographic feature of cardiac amyloidosis was thickening of the right ventricular wall, septum, and left ventricular free wall. The size of the left ventricular cavity was reduced. Approximately 20% of patients had congestive heart failure, but echocardiographic studies showed that twice as many had cardiac amyloid deposits. Patients with overt cardiac amyloidosis often presented with mild symptoms of fatigue and dyspnea on exertion. The median survival time of patients with cardiac amyloidosis and a septal thickness greater than or equal to 15 mm or less than 15 mm at diagnosis was 1 year and 4 years, respectively. Syncope with exercise developed for almost one third of patients with cardiac AL. A median survival time of 2 months is reported with exercise-induced syncope in AL.238 Immunofixation of the serum and urine and a free light-chain assay are required for any patient who presents with cardiomyopathy or congestive heart failure that does not have an obvious ischemic cause.74 The echocardiogram is the diagnostic standard for recognizing amyloid cardiomyopathy; occasionally, a patient has a nondiagnostic echocardiogram and amyloidosis is subsequently proven through endomyocardial biopsy, but these occurrences are rare.239 Pulsed tissue Doppler imaging, when combined with strain imaging, may disclose early signs of amyloidosis. However, functional abnormalities may be detected before any morphologic echocardiographic abnormalities are evident. Both the left- and right-side heart functions may be affected.240
A poor prognosis is associated with right ventricular dilatation and atrial systolic failure.241 For patients who are assumed to have hypertrophy because of thickened ventricular walls, the finding of thickened mitral and tricuspid valves is a common and important clue: thickened valves are not found in hypertensive cardiomyopathy.242 Valvular regurgitation is commonly seen by Doppler echocardiography, but it does not appear to have a clinically significant effect on myocardial performance.242 Atrioventricular sequential pacing does not benefit cardiac hemodynamics.236 Implantation of a cardioverter-defibrillator does not prevent death from cardiac amyloidosis.243 Distinguishing pericardial disease from restrictive cardiomyopathy may be difficult.244 It is rare for surgical pericardiectomy to provide clinical benefits to patients with AL. The hazards of surgery for patients with cardiac amyloidosis are well established.244, 245 As discussed previously, a bone marrow biopsy and a fat aspirate will establish the diagnosis of cardiac amyloidosis for most patients with the disease, but for those with infiltrative cardiomyopathy, endomyocardial biopsy will provide the correct diagnosis for all patients when at least three endomyocardial specimens are obtained.246 Magnetic resonance imaging may be a useful diagnostic technique. The combination of subtle, widespread heterogeneous myocardial enhancement on delayed postcontrast inversion recovery in T1-weighted images, with ancillary features of restrictive cardiac disease, may be highly suggestive of cardiac amyloidosis.247 The presence of a positive cardiac magnetic resonance imaging in AL was associated with a significantly increased risk of death, in particular of cardiac origin, but was not independent of clinical congestive heart failure.248
Ventricular thrombi may develop in AL patients because of stasis of blood within the cardiac chambers. Embolism may result, and the first manifestation of AL may be a stroke.249 For AL patients in sinus rhythm, atrial thrombi may develop.250 Anticoagulation therapy is indicated for AL patients with atrial standstill. Most AL patients present with cardiac muscle infiltration and pump failure, but patients rarely present with symptoms of exertional angina and myocardial infarction from amyloid deposition in the coronary arteries.251 Angiographic findings are normal because epicardial coronary arteries are spared. For such patients, standard exercise tests show ischemia,231 but the diagnosis of intracoronary amyloid is difficult to establish before death. Right ventricular myocardial biopsy may show amyloid deposits in small intramural vessels. We previously have described 11 AL patients who presented with angina or an unstable coronary syndrome.252 A classic low-voltage electrocardiogram was seen in only two patients. The median survival after symptom development was 18 months. Virtually all patients were assigned the diagnosis of cardiac amyloidosis during autopsy, which reflects the difficulty of recognizing small-vessel coronary arteriolar amyloidosis. A follow-up report of obstructive intramural coronary amyloidosis showed that 63 of 96 patients (66%) did not have obstruction of epicardial coronary arteries.253 Myocardial ischemia affected 25% of patients with obstructive intramural coronary amyloidosis. Most patients with primary systemic amyloidosis and cardiac involvement have obstructive intramural coronary amyloidosis and microscopic changes of myocardial ischemia.253
Not all patients with cardiac amyloid have AL. Familial amyloid cardiomyopathy, particularly in elderly African American men, must be distinguished from AL.191 The clinical presentations are similar,254 but patients with AF do not have a monoclonal protein in the serum and urine. The most common mutation identified in patients with AF in the United States is TTR Val122Ile. A screen for TTR mutations helps differentiate between patients with AF (have the mutation) and AL (do not have the mutation). Senile cardiac amyloidosis, which results from the deposition of nonmutated TTR, must also be distinguished from AL.255 Echocardiographic features of AL, AF, and senile cardiac amyloidosis are indistinguishable. All amyloid deposits are positive for Congo red stain, so positive staining cannot be used as a criterion to classify patients with different types of amyloidosis. The main distinguishing clinical feature is the presence of the monoclonal protein for patients with AL (patients with the TTR forms of cardiac amyloid lack monoclonal protein).186
Patients with senile systemic amyloidosis tend to be older than those with AL, and virtually all patients with senile systemic amyloidosis are men. Proteinuria is not present. Patients tend to have a greater left ventricular wall thickness. The severity of heart failure is less, and the median survival time is much longer than that of patients with cardiac AL.256 The mechanism of amyloid deposition in senile systemic amyloidosis is not understood. A quarter of patients older than 90 years (general patient population) have cardiac amyloid deposits. Clinicians must remember that all forms of cardiac amyloidosis are not derived from immunoglobulin light chains.188
Kidney
When a patient has free monoclonal light chains in the urine, the differential diagnosis is cryoglobulinemia,257 amyloidosis, Randall-type light-chain deposition disease,258 and myeloma cast nephropathy.259 The presence of proteinuria does not always indicate albuminuria. All patients presenting with proteinuria should have immunofixation of the urine performed during the first evaluation to exclude light-chain-associated syndromes. Kidneys are affected in 28% of AL patients. An Italian group has reported that amyloid deposits affect the kidneys for half of their patients with AL.260 For nondiabetic adults with nephrotic syndrome, amyloidosis is observed in 12% of renal biopsy specimens261; 2.5% to 2.8% of all kidney biopsy specimens contain amyloid deposits.
The serum creatinine level at AL diagnosis has prognostic value. Patients with a creatinine value less than or greater than 1.3 mg/dl had a median survival time of 25.6 or 14.9 months, respectively. Urinary protein loss did not affect survival. Patients with higher levels of albumin loss have a shorter time from diagnosis to the development of end-stage renal disease.219 The presenting serum creatinine value exceeded 2 mg/dl for 14% of patients. The median urinary protein excretion for all AL patients was 0.75 g in 24 hours, but 30% had greater than 3 g of urinary protein in 24 hours. Only 5% of AL patients had urinary protein loss in the reference range. Monoclonal light-chain proteins were more likely to be found when the urinary protein loss was high; for example, 85% of patients with urinary protein loss exceeding 1 g/day had detectable monoclonal light chains.
The ratio of patients with underlying λ clones to patients with κ clones is 5:1 among those with nephrotic-range proteinuria. Median urinary protein loss for patients with κ or λ amyloidosis is 1.1 or 4.6 g/day, respectively. The λ light chain may predispose patients to a higher level of renal involvement, but no difference in the frequency of renal failure in κ or λ amyloidosis is observed. The median survival of patients with urinary λ light chains, urinary κ light chains, or no detectable urinary light chains is 1, 2.5, and 2.5 years, respectively. Severe hypoalbuminemia is the result of nephrotic-range proteinuria.77 Diuretics typically are required to control edema, but diuretics further aggravate intravascular volume contraction, increase hypotension, and decrease renal blood flow. Bilateral catheter embolization of renal arteries reduces the loss of urinary protein and increases the serum total protein of patients with advanced anasarca.262
Continuous urinary protein loss results in tubular damage, and the principal long-term complication is end-stage renal disease.263 Prognostic factors for development of end-stage renal disease were serum creatinine value at presentation and 24-hour urinary protein loss. The median time from the diagnosis of AL nephrotic syndrome to dialysis was 14 months. After initiation of dialysis, the median survival time was 8 months. Most deaths of dialysis patients with AL were attributable to cardiac amyloid. No survival difference has been recognized between hemodialysis and peritoneal dialysis.264 The 1-year survival rate from the start of dialysis was 68%.
Only gold members can continue reading. Log In or Register to continue