Kd
References
PD-1:pembrolizumab
0.028 nM
(Hamid [89] #114) * while Kd 28 pM, 50 % effective binding concentration of pembrolizumab was 0.1–0.3 nM
PD-1:nivolumab
2.6 nM
(Brahmer [81] #48) scatchard plot analysis
PD-1:pidilizumab
20 nM
(Atkins [82] #390)
PD-1:PD-L2
89–106 nM
(Youngnak [130] #433) scatchard plot analysis
PD-1:PD-L1
270–526 nM
(Youngnak [130] #433) scatchard plot analysis
590–770 nM
(Butte [131] #8) scatchard plot analysis
B7-1:CTLA-4
400 nM
(van der Merwe [132] #432) scatchard plot analysis
B7-1:PD-L1
1,540–1,990 nM
(Butte [131] #8) scatchard plot analysis
B7-1:CD28
4,300 nM
(van der Merwe [132] #432) scatchard plot analysis
CTLA-4 was the first of the immune checkpoints to be successfully targeted in cancer. Ipilimumab (Yervoy, Bristol-Myers Squibb) is a fully human IgG1 monoclonal antibody-blocking CTLA-4, which received FDA approval in 2011 for improving overall survival in patients with metastatic melanoma [46, 47]. Patients with RCC were included in early trials with CTLA-4-blocking agents [48–50]. In a small phase 2 study of patients with advanced RCC, ipilimumab produced a 13 % response rate but with a relatively high rate (33 %) of grade 3 and 4 immune-related toxicities, such as enteritis and hypophysitis [51]. Like HD-IL-2, immune-related adverse effects have been reported to be associated with increased rates of antitumor response: ORR in patients with enterocolitis were 36 % for metastatic melanoma and 35 % for RCC, compared with 11 % and 2 %, respectively, in those without enterocolitis. [49]. While the search for predictive biomarkers has yet to produce an actionable biomarker for ipilimumab, a number of pharmacodynamic markers have been reported, such as the absolute lymphocyte count and the percentage of ICOS+ lymphocytes [52, 53]. Gene expression profiling of pretreatment tumors suggests that those with a high-baseline expression levels of immune-related genes, such as T cell markers and chemokines, are more likely to respond to ipilimumab [54].
16.5 Programmed Cell Death-1 (PD-1)
Clinically, antibodies blocking the PD-1/PD-L1 interaction have moved to the forefront of development, due to their higher activity and better tolerability than α-CTLA-4. Unlike with the CTLA-4 models, neither PD-1 nor PD-L1 knockout mice succumb to lethal autoimmunity; instead, some additional insult appears to be required for the development of overt autoimmunity [55–59]. Functionally, PD-1 is an activation marker on T lymphocytes. Activation-induced expression generally declines when antigen is cleared; this has been observed in RCC, as nephrectomy (which presumably eliminates tumor antigen) is associated with a decline in PD-1 levels on T cells [60–62]. As originally described in the lymphocytic choriomeningitis virus (LCMV) model of chronic infection, if an immune response does not successfully eliminate the antigen, prolonged antigen stimulation leads to persistent PD-1 expression [60, 61]. This high expression of PD-1 is associated with an “exhausted,” dysfunctional T cell phenotype. Viruses, bacteria, and even parasites may exploit this pathway.
Expression of the major ligand for PD-1 (PD-L1) can be induced in most cell types by the TH1 effector cytokine IFN-γ; thus, expression is generally associated with inflammation. For example, in HSV keratitis, expression of PD-L1 is upregulated on CD11b+ macrophages [63]. Schistosoma mansoni infection also results in upregulation of PD-L1 on macrophages, leading to T cell anergy [64, 65]. Homozygous PD-L1 knockout mice exhibit resistance to the parasite Leishmania mexicana [66]. As above, PD-L1 is expressed on many cell types outside of the hematopoietic lineage including cells of the epithelial lineage [35]. The generally mild phenotype of PD-L1 knockout mice, as well as its induction in the context of inflammation, suggests that PD-L1 generally serves to dampen a potentially overexuberant immune response and to prevent an anti-pathogen response from leading to autoimmunity [67]. Many tumor types express PD-L1, providing evidence that they have co-opted the PD-1/PD-L1 pathway to protect themselves from immune attack [36].
16.6 Mechanism of Immune Evasion by RCC
RCC is considered an immunogenic tumor as demonstrated by its responsiveness to IL-2 and further supported by anecdotal reports of involution of metastatic disease after removal of the primary tumor [68, 69]. Truly durable responses observed with HD-IL-2 occur in over 50 % of patients who reach a complete remission. However, complete remissions occur in less than 10 % of treated patients. Unfortunately, the majority of patients fail to achieve any benefit from HD-IL-2. Tumors can evade the immune system through both inherent and adaptive resistance to the immune system [70]. Some tumors do not appear to stimulate an active immunologic response in the tumor microenvironment, while other tumors have a lymphocyte-rich milieu. In several cancer types, an increased level of TIL can be a positive prognostic sign, such as colorectal cancer (Jass [71]). In RCC, however, higher levels of PD-L1-positive lymphocytes in tumors and PD-L1 expression on the tumor are generally associated with an increased risk of death from cancer (Fig. 16.1) [72].
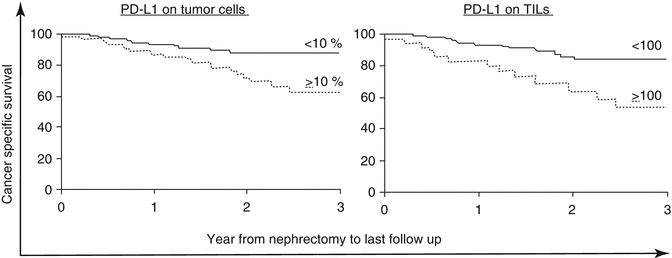
Fig. 16.1
Both PD-L1 on tumor cells and immune infiltrate are associated with death from kidney cancer (Thompson et al. [70]. Printed with permission, Copyright (2004) National Academy of Sciences, USA)
As discussed above, one potential mechanism for PD-L1 upregulation in the tumor microenvironment is the secretion of IFN-γ by effector T cells. This upregulation may be considered to be “adaptive,” in that some tumors resist T cell attack by adaptively upregulating PD-L1 as a defense mechanism. Support for “adaptive resistance” comes from recent studies in melanoma, where regions of PD-L1 expression are generally in close proximity to areas of active T cell infiltration [73]. The notion of adaptive immune resistance also explains why tumors that express PD-L1 are more likely to respond to PD-1 blockade; such tumors likely have an active, tumor-specific T cell response that is being held in check by the PD-1/PD-L1 axis, so blocking that checkpoint with appropriate antibodies relieves T cell suppression and leads to an effective T cell response, at least in some patients [74].
16.7 Differences Between PD-1 Pathway-Blocking Agents
Blocking either PD-1 or PD-L1 has shown clinical activity in patients with RCC, and there are multiple antibodies under development targeting either PD-1 or PD-L1. While anti-PD-1 antibodies all target the binding site of PD-L1 to PD-1, there may be distinct differences in their clinical benefit. Interfering with the binding site on the receptor PD-1 blocks the interaction between PD-1 and either of its ligands PD-L1 or PD-L2, but not the PD-L1:B7-1 interaction (Fig. 16.2a). Targeting the binding site on the ligand PD-L1 blocks PD-L1 binding of both PD-1 and B7-1, but not the PD-1/PD-L2 interaction (Fig. 16.2b). The different PD-1 and PD-L1 inhibitors have yet to be directly compared in clinical trials. Some have proposed that anti-PD-L1 blockade may produce fewer adverse effects than anti-PD-1 blockade. The safety of combining PD-1 and PD-L1 blockade is being tested in an ongoing phase I trial (NCT02118337).
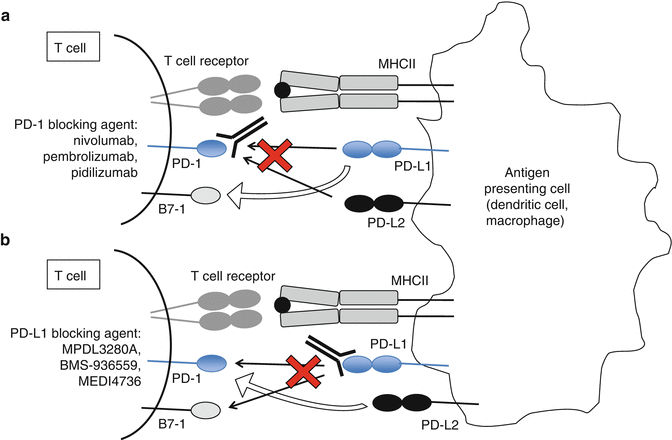
Fig. 16.2
Blocking PD-1 (a) or PD-L1 (b) may have different clinical effects, since the receptor PD-1 and the ligand PD-L1 have alternative ligands and receptors (open arrows), which are not blocked by the therapeutics listed
Variability in clinical benefit among the blocking antibodies may result from intrinsic differences in their structure and engineering. Most antineoplastic antibodies, such as trastuzumab or rituximab, mediate their effects through antibody-dependent cellular cytotoxicity (ADCC) [75]. However, the primary mechanism by which immune checkpoint-targeted antibodies function is by physically blocking the binding of the receptor and the ligand, not by the destruction or depletion of lymphocytes. Indeed, the expression of PD-1 on activated as well as “exhausted” lymphocytes would make it seem unwise to generate a depleting anti-PD-1 antibody, although to our knowledge a PD-1 antibody optimized for depletion has not yet been evaluated in preclinical studies. So, aside from pidilizumab/CT-011, which is an IgG1 isotype antibody, all other PD-L1 and PD-1-blocking antibodies are engineered antibodies with mutated ADCC-activating sites in the Fc domain, or IgG2 or IgG4 antibodies, which have minimal ADCC/CDC activity (Table 16.2).
Table 16.2
Summary of the function and isotype of PD-1 and PD-L1-blocking antibodies currently in development
Function | Killer isotype | Non-killer isotype | |
|---|---|---|---|
Anti-CTLA4 | Blocks the interaction between CTLA-4 and its receptors B7-1 and B7-2 | Ipilimumab (IgG1) | Tremelimumab (IgG2) |
Anti-PD-1 | Blocks the interaction between PD-1 and its ligands PD-L1 and PD-L2 | Pidilizumab (IgG1) | Nivolumab (IgG4) |
Pembrolizumab (IgG4) | |||
Anti-PD-L1 | Blocks the interaction between PD-L1 and its receptors PD-1 and CD80 | BMS-936559 (IgG4) | |
MPDL3280A (mutated IgG1a) | |||
MEDI4736 (engineered IgG1) |
Indeed, a better understanding of the biology of immune checkpoint blockade may enhance our knowledge about the mechanisms underlying the clinical effects. For example, the checkpoint inhibitors ipilimumab and tremelimumab both block CTLA-4:B7 through binding CTLA-4 on lymphocytes. Like ipilimumab, tremelimumab elicited promising efficacy in early phase I and II studies [76, 77]. Unfortunately, the phase III trial of tremelimumab in melanoma showed no significant difference in response rate or overall survival over standard of care chemotherapy [78]. Clinically, the mechanism(s) underlying the antitumor effect of ipilimumab is not fully understood. As mentioned above, recent preclinical studies indicate that the ADCC-activating (killer) Fc domain is necessary for the antitumor effect of anti-CTLA-4 and that a portion of its antitumor efficacy is driven by depletion of T regulatory cells [44, 45]. While CTLA-4 is only transiently expressed on most T cells, T regulatory cells constitutively express CTLA-4. Higher levels of CTLA-4 expression have been found on Tregs in the tumor compared to effector T cells [79]. Interestingly, tremelimumab is an IgG2 isotype (non-killer) antibody, while ipilimumab is an IgG1 isotype (killer) antibody (Table 16.2).
The available PD-1 and PD-L1 antibodies may also vary in their affinity of the antibody for its target. Since all antibodies target the PD-1/PD-L1 binding region, there is probably relatively little difference between the antigen targets of the antibodies. However, the breadth of difference in antibody affinity of three anti-PD-1 antibodies is illustrated in Table 16.1 [80–82]. Of the PD-1-directed antibodies, pembrolizumab has been reported to have the highest affinity for PD-1 (Kd 20 pM), although it is challenging to compare affinities of different antibodies when the assays used to quantify affinity are not uniform. Nonetheless, a higher affinity agent may be a more effective blocking agent and would also allow an antibody to remain efficacious at lower concentrations observed long after administration or at sites with low antibody penetrance.
16.8 Early Studies: Efficacy and Toxicity
16.8.1 Anti-PD-1 Blockade in RCC
16.8.1.1 Nivolumab
Nivolumab is an IgG4 antibody against PD-1 and has been approved in Japan for the treatment of melanoma. The first in-human trial of an anti-PD-1 antibody as a monotherapy treatment in RCC was a pilot phase I, dose-escalation study (BMS-936558, MDX-1106, ONO-4538; Bristol Meyers Squibb). One RCC patient was included; that patient achieved a partial response during the study period [81], but he then went on to develop a long-term complete response (CR) after receiving only three doses of study drug (C. Drake, personal communication). A larger, dose-escalation phase I/II trial of nivolumab included 34 patients with RCC, who were treated every 2 weeks for up to 96 weeks at doses ranging from 0.1 to 10mg/kg [83]. In that phase Ib study, a maximum tolerated dose was not reached in dosages up to 10 mg/kg. Five percent of patients stopped treatment due to toxicity. The immune-related toxicity seen in this study included pneumonitis, vitiligo, colitis, hepatitis, hypophysitis, and thyroiditis and was not correlated with dose level. Endocrinopathies were generally effectively managed with replacement doses of appropriate hormones. Severe cases of colitis and transaminitis were reversed with treatment interruption or corticosteroids. Pneumonitis was observed in 3 % of all solid tumor patients (9/296). Three cases of pneumonitis-related death were reported (none in RCC patients). These cases prompted strict vigilance for this life-threatening toxicity as well as prompt administration of corticosteroids for clinically significant immune-related adverse events. This enhanced awareness for this adverse event may explain the lack of additional deaths secondary to pneumonitis in subsequent studies. Other adverse effects included fatigue, rash, pruritus, nausea, and decreased appetite. Ten of the 34 patients with heavily pretreated RCC experienced objective responses to nivolumab [84]. Subsequent reporting revealed an encouraging median PFS of 7.3 months and a median OS at 22 months [85].
Two early phase trials have been reported in RCC. A phase II dose-ranging study enrolled 168 patients with RCC with a clear cell component and prior antiangiogenic therapy (NCT01354431) [86]. The type and pattern of adverse effects was similar to previously reported studies. There were no grade 3–4 pneumonitis or grade 5 events reported. Overall response rates were 20–22 % across three dose cohorts. PFS ranged 2.7–4.2 months. OS was perhaps the most encouraging result at 18–25 months. A concurrent phase 1 biomarker trial enrolled 67 patients with clear cell mRCC, who had received 1–3 prior treatments, as well as 24 treatment-naive patients (NCT01358721) [87]. The overall response rate was 17 %. In the previously treated cohort, patients who received the higher dosing of nivolumab at 10 mg/kg had an objective response rate of 22 %. In the untreated patients who were also treated with nivolumab 10 mg/kg, the response rate was 13 %. While these are very small cohorts, the finding of a lower ORR in the treatment-naive cohort was somewhat surprising. A large, phase III randomized trial has accrued and will provide definitive data as to whether nivolumab increases overall survival compared to everolimus in a treatment refractory setting (NCT01668784). Additional studies are ongoing to evaluate whether there is a role for PD-1 pathway inhibitors in the first-line setting of RCC or whether prior VEGF-directed treatment of RCC increases the response rates to PD-L1 blockade.
16.8.2 Anti-PD-L1 Blockade in RCC
16.8.2.1 BMS-936559
The first published clinical trial of PD-L1 blockade reported the initial safety and efficacy of a fully human IgG4-blocking monoclonal antibody against PD-L1, BMS-936559 (Bristol Meyers Squibb) [88]. In this phase I dose-escalation trial (NCT00729664), a maximum tolerated dose was not found, and the maximum dose administered was 10 mg/kg. Immune-mediated adverse events were observed in 39 % of patients, including rash and hypothyroidism, as well as individual cases of sarcoidosis, endophthalmitis, diabetes mellitus, and myasthenia gravis. Six percent of patients discontinued treatment due to adverse events. Of note, there were no cases of pneumonitis with this anti-PD-L1 antibody. In the subset of patients with renal cell carcinoma, only 2 of 17 patients had objective responses.
16.8.2.2 MPDL3280A
MPDL3280A (Roche/Genentech) is an engineered IgG1 monoclonal antibody directed against PD-L1. This agent has been found to be well tolerated in doses ranging up to 20 mg/kg [89]. In an expansion arm of the phase I study, 15 % of patients with advanced RCC had objective responses with a 24-week PFS rate of 51 % (95 % CI: 38–63) [90]. While most immune checkpoint trials required RCC with a clear cell component, the expansion cohort of MPDL320A also enrolled patients with non-clear cell RCC (nccRCC). Of the six nccRCC patients, one had a partial response (Table 16.3) [91]. Further investigation is warranted in patients with nccRCC as there are few effective treatment options for this subgroup. The clinical benefit of MPDL3280A has also been reported in multiple tumor types and in particular for urothelial carcinoma (UC), which has led to the FDA breakthrough designation for UC patients with PD-L1-positive tumors [92].
Table 16.3
PD-L1 blockade can benefit patients with both clear and non-clear cell RCC, as demonstrated in the phase 1b trial with MPDL3280A (Cho et al. [91])
n | ORR (%) | 24-week PFS (%) | |
|---|---|---|---|
All tumors | 140 | 21 | 45 |
RCC | 47 | 13 | 53 |
Clear cell RCC | 40 | 13 | 57 |
Non-clear cell RCC | 6 | 17 | 20a |
16.8.3 Other PD-1 Pathway-Blocking Therapies in Development in RCC
16.8.3.1 Pembrolizumab
Pembrolizumab (MK-3475, Merck) is a humanized anti-PD-1 IgG4 isotype antibody with a high affinity to PD-1 (Table 16.1). It has not yet been evaluated in patients with RCC but is the first PD-1 pathway-blocking antibody to gain FDA approval for patients with ipilimumab-refractory melanoma. Given the excellent efficacy and general tolerability observed in melanoma [80, 93], there are several ongoing clinical trials in which pembrolizumab is being combined with tyrosine kinase inhibitor (TKI) in RCC patients. These include combination with pazopanib (NCT02014636) as well as with axitinib (NCT02133742).
There are also several additional immune checkpoint-blocking antibodies in earlier stages of clinical development. These include MEDI4736, an engineered IgG1 PD-L1 antibody. Preliminary phase I data on this agent were recently reported [94] and included a single patient with RCC who responded for over 36 weeks. Dose-expansion cohorts of MEDI4736 are being studied in multiple other solid tumor types. Pidilizumab (CT-011, CureTech) is a humanized anti-PD-1 IgG1 isotype antibody, which was first tested in patients with hematologic malignancy and was found to be safe and well tolerated at doses ranging from 0.2 to 6 mg/kg [95]. However, a phase II trial in melanoma reported a relatively low overall response rate (approximately 6 %), which is somewhat inconsistent with other PD-1/PD-L1-blocking agents [82]. An ongoing phase II trial is currently assessing the safety of CT-011 alone or in combination with a dendritic cell/RCC cell fusion vaccine (NCT01441765). AMP-224 (Amplimmune and GlaxoSmithKline [GSK]) employs a different strategy for blockade; this agent is a B7-DC immunoglobulin fusion protein, which acts by binding and blocking PD-1. This agent is currently undergoing phase I investigation (NCT01352884).
16.9 Role of PD-L1 Expression in RCC
In several preclinical models, induced expression of PD-L1 on tumors increased tumorigenesis and invasiveness in vivo [15]. In addition to tumor cells, PD-L1 is expressed on infiltrating lymphocytes, monocytes, and macrophages (Fig. 16.3) [72]. In some types of cancers such as ovarian, lung, and RCC, tumoral PD-L1 expression is associated with worse prognosis [72, 96, 97]. In a retrospective cohort of patients with RCC, PD-L1 expression on both tumor cells and immune infiltrate was associated with a relatively poor prognosis (Fig. 16.1) [72]. Patients whose kidney tumors had ≥10 % tumor cell expression had a threefold increased risk of dying of their disease. The majority of the patients in this cohort had early-stage resected disease. In a retrospective analysis of the tumors from patients with metastatic disease on the phase III COMPARZ trial comparing sunitinib and pazopanib, PD-L1 expression ≥5 % was associated with worse overall survival [98]. An interesting recent study evaluated the expression of PD-L1 on tumor cells and tumor-infiltrating immune cells. These data showed that expression of PD-L1 on infiltrating immune cells varied greatly between melanoma, NSCLC, and RCC and that PD-L1 expression on tumor cells (rather than infiltrating immune cells) had the strongest association with response to nivolumab [99]. These data seem to be contradictory to those reported for the anti-PD-L1 antibody MPDL3280A, where PD-L1 expression on immune cells within the tumor appears to associate most strongly with response [89, 100–101]; however, the two assays employ different detection antibodies and likely different staining protocols, which might explain this discrepancy.
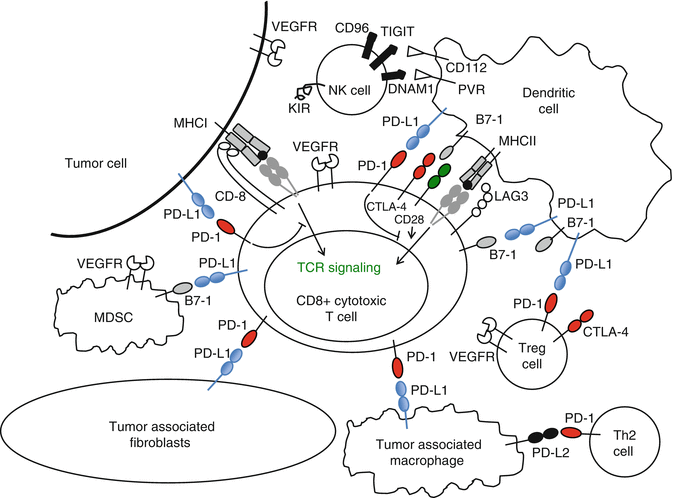
Fig. 16.3
PD-L1 is expressed on many different cells within the immune system. Checkpoint inhibitors may affect other immune cells, in addition to CD8+ effector T lymphocytes in the tumor microenvironment
In the phase II study of nivolumab in RCC, higher PD-L1 expression on tumors was associated with an increased likelihood of response, with response rates of 31 % in PD-L1-positive and 18 % in PD-L1-negative tumors (Table 16.4) [102]. It is important to note that the predictive value of PD-L1 expression with nivolumab treatment may depend on the antibody clone employed. For example, using an automated assay with the 28-8 antibody clone, the response rate in PD-L1-positive tumors was much lower across tumor types [72, 102–104] than reported in the original study with clone 5H1 [81]. With MPDL3280A, 20 % of RCC patients with PD-L1-positive tumors responded to therapy as compared to 10 % for PD-L1-negative tumors (Table 16.4) [90].
Table 16.4
Summary of clinical outcomes and PD-L1 expression in the development of PD-1 pathway blockade in RCC
Agent(s) (year reported) | Tumor type | n | Anti-PD-L1 clone | RR (%) (95 % CI) [n/N] | Pattern defined as positive | RR (%) PD-L1 positive (95 % CI) [n/N] | RR (%) PD-L1 negative (95 % CI) [n/N] | mPFS PD-L1 positive (95 % CI) | mPFS PD-L1 negative (95 % CI) | mOS (m) PD-L1 positive (95 % CI) | mOS (m) PD-L1 negative (95 % CI) |
|---|---|---|---|---|---|---|---|---|---|---|---|
Nivolumab1 2012 | Solid tumorsa | 42 | 5H1 | 40 %a | Tumor cell >5 % | 36 % | 0 % | nr | nr | nr | nr |
MPDL3280A2 2014 | Kidney cancer | 62 | PDL1dxb | 14 % | Immune infiltrate >1 % | 20 % (9–37) | 10 % (2–30) | 24 weeks (5–98+) | 20 weeks (5–94+) | nr | nr |
Nivolumab3 2014 | Kidney cancer | 107 | 28–8 | 20–22 %c | Tumor cell >5 % | 31 % (15.3–50.8) | 18 % (10.2–28.3) | 4.9 m (1.4–7.8) | 2.9 m (2.1–4.2) | NA (13.4 m-NA) | 18.2 m (10.2–28.3) |
Nivolumab/Ipilimumab4 2014 | Kidney cancer | 36 | 28–8 | 43 % [9] – 48 %[11]d | Tumor cell >5 %—>1 % | 25 % [1/4]—50 % [8/16] | 56.3 % [8/16]—55 % [11/20] | nr | nr | nr | nr |
Nivolumab/VEGF-TKI5 2014 | Kidney cancer | 33 | 28–8 | 52 %[17]e | Tumor cell >5 %—>1 % | 20 % [1/5]—40 % [6/15] | 58.3 % [14/24]—64.3 % [9/14] | nr | nr | nr | nr |
In general, higher PD-L1 expression on tumor cell and immune-infiltrating cells appears to be associated with a higher likelihood of response. However, the use of PD-L1 expression as an up-front selection biomarker is suboptimal, as a significant percentage of “negative” tumors may respond (reviewed in [105]). Furthermore, there are many patients that are “positive” who do not respond. One way in which PD-L1 expression may be useful as a biomarker is for guiding the sequence of therapy or whether monotherapy or combination therapy may be more appropriate—but those questions clearly require prospective investigation. PD-L1-based patient selection is also limited by the lack of a standardized definition of PD-L1 positivity or assay. Other entities (Genentech/Roche, Merck, and MedImmune) who are developing blocking antibodies against either PD-1 or PD-L1 also have developed distinct companion assays for PD-L1 expression each with different anti-PD-L1 antibodies. These assays have yet to be directly compared. Thus, it is impossible to determine whether the differences in correlations found between tumors and treatments reported are a function of the nature of the patient samples analyzed, the biologic differences of anti-PD-1 versus anti-PD-L1 therapy, or the assay (antibody specificity) itself.
In addition to discrepancies in the assays used, there are many other factors that may complicate the use of tumoral PD-L1 expression as a potential predictive biomarker. Tumor heterogeneity is a significant issue in many tumors, and thus, sampling bias may affect the results [106]. Further, PD-L1 is a dynamic marker that can be upregulated locally in response to cytokines induced by inflammation (adaptive resistance) [35] and in response to the selection pressures of treatment. Thus, the expression of PD-L1 within tumors and its microenvironment may change over time. Therefore the primary tumor expression may not reflect that of the metastases or even the primary tumor’s level at a later time [107]. As an example of treatment-induced changes, three similar neoadjuvant phase II trials studied the effects of the VEGF-TKIs sunitinib and pazopanib on the primary tumor and found a reduction in vessel density, PD-L1 expression, and FOXP3 expression, but increases in Fuhrman grade and Ki-67 levels [105]. In addition, PD-L1 can be induced by some oncogenic mutations such as in PTEN (rare in RCC) or directly by gene amplification [109, 107]. Once again, despite these factors, across multiple tumor types using different therapies and varied assays for PD-L1 expression, there is a clear trend for PD-L1-positive tumors to be more likely to respond to blocking antibodies than PD-L1-negative tumors [72, 89, 93, 94, 100–101, 103, 104, 111–113]. It is interesting that preliminary data suggest that PD-L1 expression on RCC tumor cells is also associated with increased clinical benefit from HD-IL-2 [114]. Thus, PD-L1 status could also play a role in building a better predictive model for optimal HD-IL-2 candidates.
16.10 Increasing Response Rates with Combination Therapies
16.10.1 Nivolumab + Ipilimumab
Several trials are underway in which PD-1 pathway blockade is combined with either novel or FDA-approved agents in an effort to build on the clinical efficacy of monotherapy (Table 16.4). Combining PD-1 inhibition with CTLA-4 blockade is one logical approach given preclinical melanoma models showing enhanced efficacy for concurrent combination treatment [115, 116]. The first phase I study combining nivolumab and ipilimumab was performed in advanced melanoma—where encouraging efficacy was observed with an impressive objective response rate of 53 % albeit at the expense of increased toxicity [117].
Related posts:
 Genetic Heterogeneity of Kidney Cancer
Angiogenesis Inhibitor Therapy in Renal Cell Cancer
Presurgical Therapy in Renal Cell Carcinoma
Genetic Heterogeneity of Kidney Cancer
Angiogenesis Inhibitor Therapy in Renal Cell Cancer
Presurgical Therapy in Renal Cell Carcinoma
 Cytokines in the Management of Advanced Renal Cell Cancer
Cytokines in the Management of Advanced Renal Cell Cancer
 Renal Cell Carcinoma: Clinical Presentation, Staging, and Prognostic Factors
Renal Cell Carcinoma: Clinical Presentation, Staging, and Prognostic Factors
 Hereditary Kidney Cancer Syndromes
Hereditary Kidney Cancer Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


