Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .


Introduction
Although severe hypertension is uncommon among patients with thyroid disease and primary hyperparathyroidism, these endocrinopathies are identified as causes for endocrine hypertension. Free thyroid hormones are positively correlated to elevated blood pressure in subjects with normal thyroid function [ ]. The three major determinants of hypertension are cardiac output, peripheral vascular resistance, and renal function. Thyroid hormones play a vital pathophysiological role in the development of hypertension by influencing the above three determinants [ ]. Both overt and subclinical stages of thyroid dysfunction—hypothyroidism and hyperthyroidism—can be associated with hypertension [ ]. Whereas hyperthyroidism is associated with elevated systolic blood pressure, hypothyroidism is associated with elevated diastolic blood pressure [ ]. Primary hyperparathyroidism—both normocalcemic as well as hypercalcemic—is also associated with hypertension, and parathyroidectomy may be associated with improvement in systolic blood pressure and diastolic blood pressure, as well as normalization of biochemical parameters [ , ].
Thyroid disease
Physiology of hypothalamo–pituitary–thyroid axis and cardiovascular system
In response to thyroid stimulating hormone (TSH), the thyroid gland synthesizes, stores, and secretes two biologically active thyroid hormones: thyroxine (T4) and tri-iodothyronine (T3). Once released, 80% of the circulating T3 is derived from peripheral deiodination of T4. Free thyroid hormone levels exert direct negative feedback on the synthesis and secretion of the pituitary TSH, and an indirect negative effect on the synthesis and secretion of thyrotropin-releasing hormone through inhibition at the level of hypothalamus. Iodine is an essential component of the thyroid hormones; four iodine atoms and three iodine atoms are added to thyronines bound to thyroglobulin to create T4 and T3, respectively. Thyroid hormone is essential for the normal functioning of every cell, and in particular it influences protein, carbohydrate
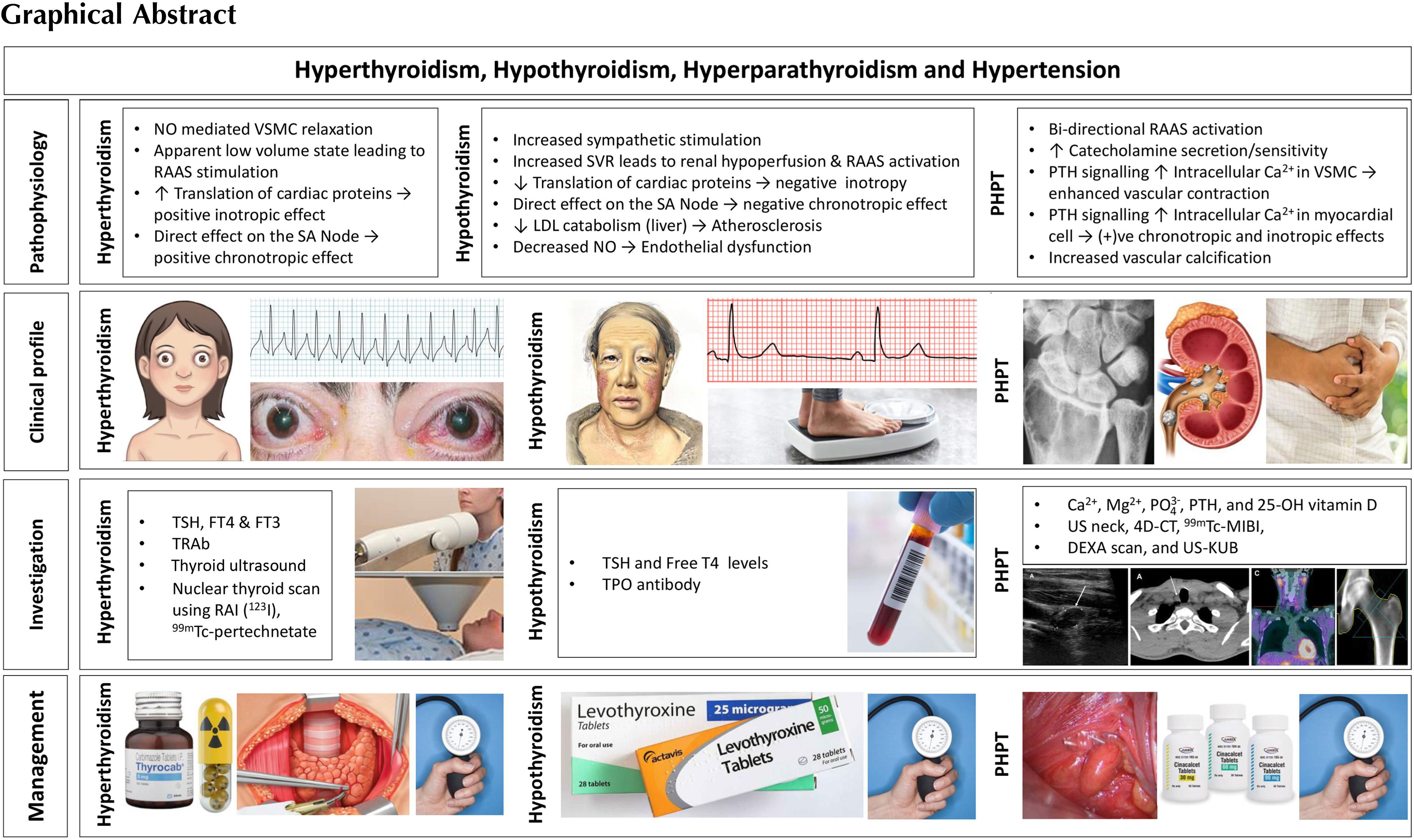
Activation of cardiac-specific thyroid hormone receptors control the translation of various cardiac proteins including the sarcoplasmic reticulum calcium adenosine triphosphatase (SERCA or SR Ca 2+ -ATPase), phospholamban, α-myosin heavy chain (α-MHC), β-myosin heavy chain (β-MHC), β1-adrenoceptors, atrial natriuretic hormone, sodium/potassium-ATPase (Na + /K + -ATPase), sodium-calcium exchanger (NCX), and voltage-gated potassium channels [ , , ]. Excess or reduced thyroid hormone availability can, therefore, result in distinct changes in the production of these cardiac proteins leading to alterations in the numerous cardiovascular parameters they control.
Pathophysiology of hypertension in thyroid dysfunction
Blood pressure is regulated by cardiac output and systemic vascular resistance [ ]. In healthy individuals, T3 activity reduces the vascular resistance, increases the cardiac contractility and chronotropy, and also alters the sensitivity to sympathetic stimulation [ , , , ]. Therefore, multiple systemic changes seen in hypo- and hyperthyroidism contribute to alterations in blood pressure.
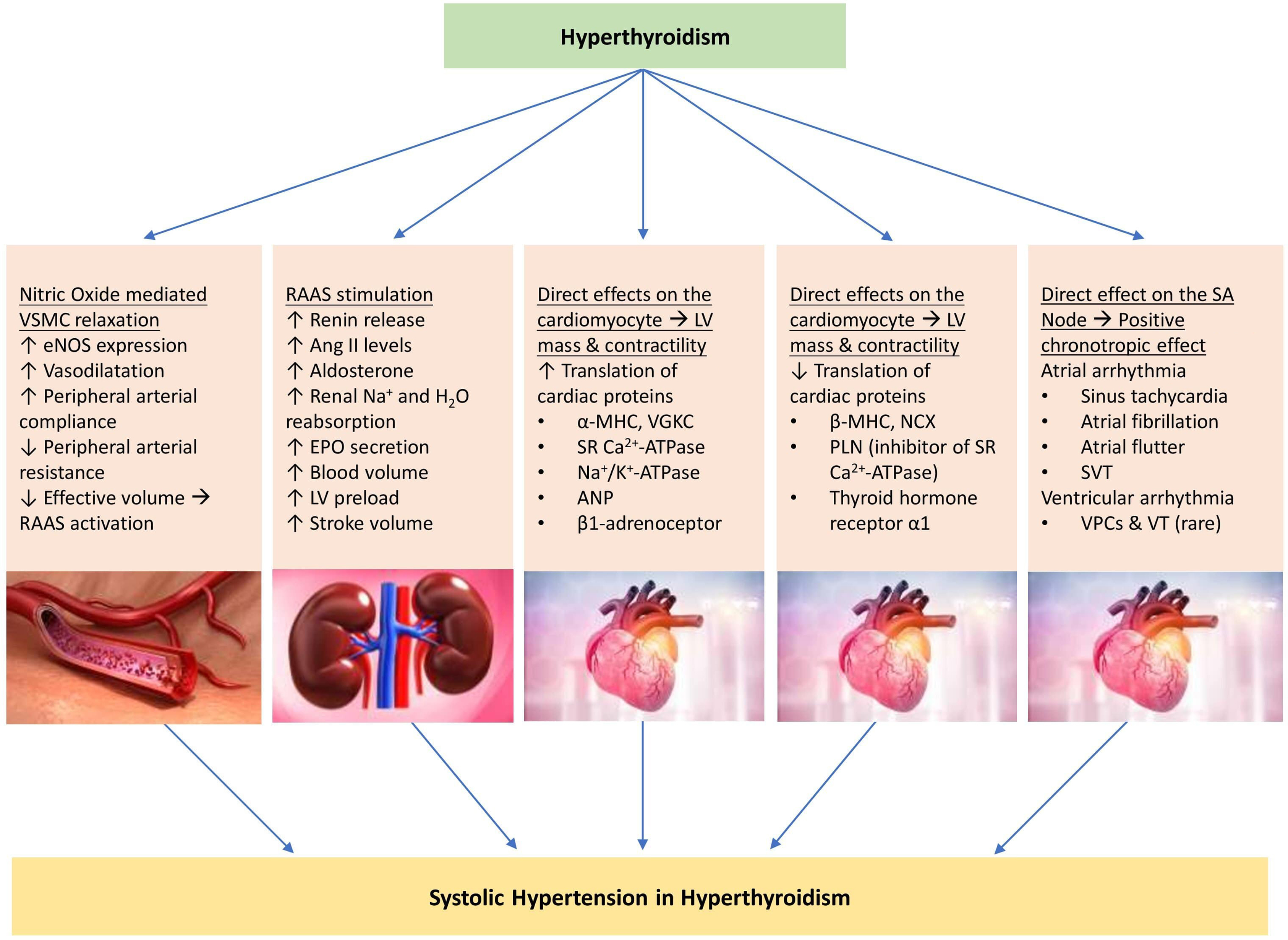
Hyperthyroidism decreases the systemic vascular resistance due to nitric oxide-mediated vascular smooth muscle relaxation [ , , ]. It also increases the cardiac contractility [ , , , ] and heart rate [ , , ]. Stimulation of increased protein synthesis in cardiac myocytes in response to increased hemodynamic load (from increased contractility and heart rate) can lead to left ventricular hypertrophy, resulting in impaired diastolic filling [ , , ]. As a result of decreased systemic vascular resistance, there is an apparent low-volume state, stimulating the release of renin and aldosterone. This stimulates sodium reabsorption from the kidney, increasing the blood volume and the resultant cardiac output [ , , ]. Hyperthyroidism preferentially elevates the systolic blood pressure with a resultant widened pulse pressure [ , , , ]. Hyperthyroidism is also associated with pulmonary hypertension. It is postulated that the decreased systemic vascular resistance usually observed in response to excess T3 does not occur in the pulmonary vasculature. In association with a high cardiac output state, this can lead to right heart failure resulting in pulmonary hypertension [ ]. The cardiac response to subclinical hyperthyroidism is less uniform. However, prolonged exposure to subclinical thyroid hormone excess can lead to increased heart rate, arrhythmias, and left ventricular hypertrophy, the common cardiac manifestations that can contribute to elevations in blood pressure [ , , ]. The pathophysiological mechanisms leading to the development of hypertension in hyperthyroidism are depicted in Fig. 16.1 .
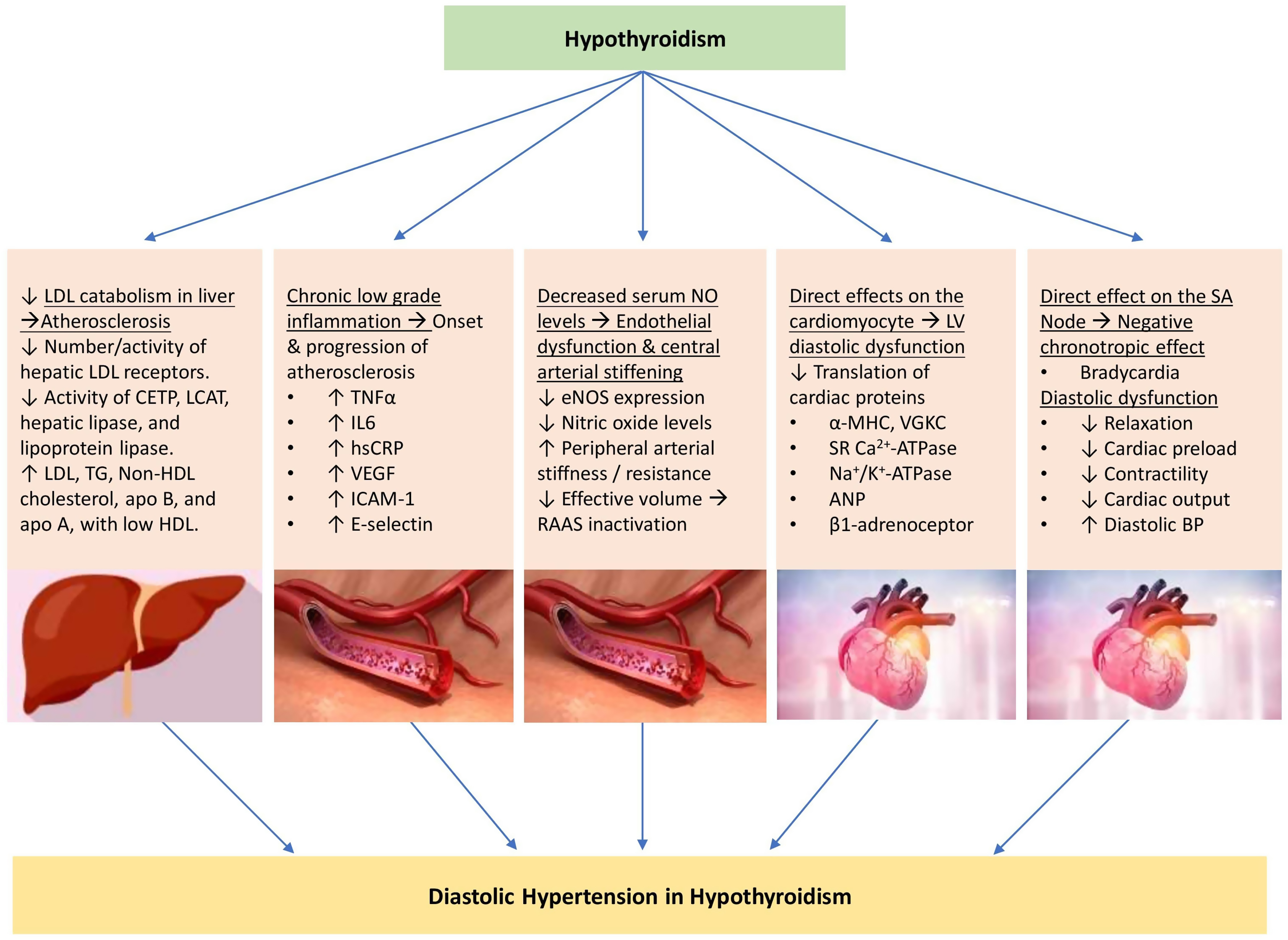
Hypothyroidism results in increased systemic vascular resistance due to lowered arterial compliance [ , , , , ], decreased cardiac contractility [ ], decreased heart rate, cardiac output, and blood volume [ , , , ], and is associated with delayed ventricular diastolic relaxation [ ]. In addition to left ventricular diastolic dysfunction, hypothyroidism has also been demonstrated to compromise longitudinal left ventricular systolic function [ ]. Hypothyroidism is also associated with increased sympathetic stimulation and elevated catecholamine levels [ , ]. Increased systemic vascular resistance leads to renal hypoperfusion and reduced glomerular hydrostatic pressure. This results in activation of the macula densa and renin–angiotensin–aldosterone system (RAAS) stimulating further vasoconstriction and resulting in decreased total body water excretion [ , , ]. In addition, a lack of regular antidiuretic hormone (ADH) suppression in the absence of sufficient thyroid hormone also leads to further total body water retention [ , ]. Weight gain associated with hypothyroidism may also contribute to the increased risk of hypertension [ ]. Compared with hyperthyroidism, hypothyroidism preferentially increases diastolic blood pressure and results in a narrowed pulse pressure [ , , , ]. The evidence for hypertension associated with subclinical hypothyroidism is mixed. However, prolonged exposure to an elevated TSH is associated with elevation of surrogate cardiovascular risk factors, such as increased carotid intima-media thickness, and therefore is also likely to contribute to the cardiovascular alterations and hypertension [ , ].
The pathophysiological mechanisms associated with the development of hypertension in subjects with hypothyroidism are shown in Fig. 16.2 .
Subclinical or overt hypothyroidism and associated hypertension
Epidemiology
Overt primary hypothyroidism is defined as an elevated TSH level and a low free T4 level, while subclinical hypothyroidism is defined as an elevated TSH level with a normal range free T4 level [ ]. Secondary hypothyroidism will involve a low level of free T4, but with a normal or low level of TSH. The prevalence of overt hypothyroidism in the population ranges from 0.2% to 5.3%. Hypothyroidism preferentially affects women, its prevalence increases with advancing age, and is more common in Caucasian populations [ ]. The prevalence of subclinical hypothyroidism ranges from 3.4% to 9.0% [ , ]. The prevalence of hypertension in patients with hypothyroidism varies widely with a reported range of 15%–43% [ ]. The causes of primary hypothyroidism include chronic autoimmune lymphocytic thyroiditis (Hashimoto’s), infectious or infiltrative thyroiditis (e.g., IgG4-associated, amyloidosis, hemochromatosis), iodine deficiency, radioactive iodine (RAI) therapy, thyroidectomy, external beam neck irradiation, and various drugs (e.g., lithium, amiodarone, and immune checkpoint inhibitors, i.e., CTLA4 inhibitors and PD-1 inhibitors). Secondary or central hypothyroidism results from various hypothalamic or pituitary diseases [ , , , ].
Clinical features
Symptoms of hypothyroidism are related to the severity and duration of hypothyroidism and include constipation, fatigue, weight gain, cold intolerance, muscle weakness and cramps, exertional dyspnea, changes in the menstrual cycle, cognitive dysfunction, and impaired memory [ ]. Clinical features may include goiter, dry skin and hair, periorbital edema, hair loss, macroglossia, and delayed tendon reflex relaxation. Atrioventricular block, sinus bradycardia, prolonged QTc interval with the rare potential for torsades de pointes, and pleural and pericardial effusions are all features associated with hypothyroidism [ , ]. Diastolic hypertension is also a common feature. Moreover, hypothyroidism is associated with elevated cholesterol levels, in particular a marked increase in low-density lipoproteins (LDLs) and apolipoprotein B, mainly due to the reduced hepatic LDL clearance. This contributes to accelerated atherosclerosis, and when combined with additional coagulation protein changes, there is a significantly increased risk of coronary artery disease [ , , , ]. Excess free water retention can lead to dilutional hyponatremia associated with an elevated ADH level [ ]. Severe hypothyroidism can result in myxedema coma, which is identified by decreased conscious state with significant hypothermia, bradycardia, hypoventilation, and hyponatremia. Clinical findings are subtle in subclinical hypothyroidism compared with overt hypothyroidism, with the majority of individuals remaining asymptomatic [ , ].
Investigation and management
Laboratory diagnosis of hypothyroidism requires measurement of serum TSH and T4 levels. Because autoimmune disease is a common cause, serum antibodies directed against thyroid peroxidase (TPO) and thyroglobulin are often elevated [ ]. Management of hypothyroidism generally involves thyroid hormone replacement with levothyroxine. In primary hypothyroidism, levothyroxine dosing is adjusted to achieve a TSH level within the normal laboratory reference range which results in resolution of signs and symptoms of hypothyroidism. In secondary hypothyroidism, the dose of levothyroxine should be adjusted to achieve a mid- to high-normal free T4. The use of liothyronine (T3) is not routinely required in the vast majority of patients with hypothyroidism [ , ].
Impact of treatment of hypothyroidism on hypertension and cardiovascular disease
Treatment of hypothyroidism with exogenous levothyroxine administration reduces the systemic vascular resistance [ ] as well as the other cardiovascular alterations seen in hypothyroidism [ , ]. Overall, this leads to improvements in blood pressure, and hypertension can abate without the need for additional antihypertensive agents [ , , ]. The treatment benefit of subclinical hypothyroidism is not as well established, but there is evidence that levothyroxine replacement can improve the blood pressure control, and therefore overall cardiovascular risk reduction [ ].
Subclinical or overt hyperthyroidism and associated hypertension
Epidemiology
Overt hyperthyroidism is defined as a low, usually suppressed, TSH level and an elevated T4 and/or T3 level. Subclinical hyperthyroidism is defined as a low TSH level with a normal T4 level, without interference from hypothalamic, pituitary, or nonthyroidal illness [ , ]. The prevalence of overt hyperthyroidism ranges from 0.1% to 0.5% of the population, and this preferentially affects women and non-White populations [ , , ]. The prevalence of subclinical hyperthyroidism ranges from 0.5% to 2.1% [ , ]. The prevalence of hypertension in hyperthyroid patients is 20%–30%, but can be above 50% in those aged over 60 years [ ]. The prevalence of pulmonary hypertension in patients with hyperthyroidism can range from 36% to 65%, though the majority of cases are mild [ ]. The common causes of hyperthyroidism are Graves’ disease, toxic multinodular goiter (MNG), autonomously functioning toxic nodules, and subacute thyroiditis. Excessive iodine intake or exposure can provoke hyperthyroidism if an underlying thyroid disease is present (e.g., euthyroid MNG). Amiodarone, as a source of excess iodine, can cause hyperthyroidism as in type 1 amiodarone-induced hyperthyroidism but is also a source of organified iodine that can cause thyroiditis as in type 2 amiodarone-induced hyperthyroidism. Excessive thyroid hormone replacement can cause factitious or iatrogenic hyperthyroidism [ , , ].
Clinical features
Symptoms of hyperthyroidism include weight loss despite normal or increased appetite, fatigue, palpitations, exertional dyspnea, tremor, heat intolerance, muscle weakness, diplopia, anxiety, agitation, poor concentration, disrupted sleep, increased stool frequency, and changes in the menstrual cycle, usually oligomenorrhea [ , , ]. Clinical features include hyperactivity, fine tremor, warm skin with increased perspiration, hair thinning, proximal myopathy, hyperreflexia, and the eye signs of lid retraction and lid lag. In Graves’ disease, autoimmune thyroid eye disease can cause proptosis, conjunctival injection, and ophthalmoplegia, and autoimmune dermopathy usually manifests as pretibial myxedema [ , , ]. Tachycardia is a common finding with an increased risk of atrial arrhythmias (atrial fibrillation and premature atrial complexes), as well as left ventricular hypertrophy, and heart failure [ , , , , , ]. Although hyperthyroidism does not adversely affect lipid metabolism, it can still lead to elevated coronary artery disease risk due to endothelial dysfunction [ ]. Hyperthyroidism can cause hypercalcemia due to increased bone turnover, leading to decreased bone mineral density, and increased fracture risk [ , , ]. A rare complication of hyperthyroidism is hypokalemic thyrotoxic periodic paralysis caused by acute potassium shifts into muscle cells due to mutations in the thyroid hormone-regulated potassium channels, most frequently seen in persons of Chinese ethnicity [ ].
Investigation and management
Laboratory diagnosis of hyperthyroidism involves measurement of serum TSH, free T4, and free T3 levels. Antibodies against the TSH receptor (TRAb) are a hallmark of Graves’ hyperthyroidism. The ratio of serum T3 to T4 may also indicate the underlying etiology of hyperthyroidism; a hyperactive thyroid gland will produce more T3 than T4, while a destructive thyroiditis will release more preformed T4 than T3. Nuclear thyroid imaging, usually with a technetium ( 99m Tc) scan, can assist in determining the etiology of hyperthyroidism. Diffusely increased (homogenous) uptake is indicative of Graves’ disease, focal uptake with suppressed uptake in the remainder of the thyroid indicates a toxic adenoma (autonomously functioning thyroid nodule), a toxic MNG is represented by multiple focal areas of increased and reduced uptake (nodular heterogenous uptake), while diffusely reduced or absent uptake indicates a destructive process such as subacute thyroiditis, postpartum thyroiditis, iodine overload, or factitious hyperthyroidism from exogenous thyroid hormone. Thyroid ultrasonography with color flow Doppler imaging can determine the presence of structural abnormalities as well as changes in thyroid vascularity. Increased vascularity is more consistent with a hyperactive gland (e.g., Graves’ hyperthyroidism and type 1 amiodarone-induced hyperthyroidism), while decreased vascularity suggests a destructive thyroiditis (e.g., type 2 amiodarone-induced hyperthyroidism) [ , ].
Management of hyperthyroidism, irrespective of the underlying etiology, involves both acute interventions to render a patient euthyroid, as well as long-term definitive options to prevent further recurrences. Destructive thyroiditis is managed with ongoing monitoring, beta-adrenergic blockade for symptom benefit, and nonsteroidal antiinflammatory drugs, or glucocorticoids if required. Glucocorticoids are often necessary for type 2 amiodarone-induced hyperthyroidism. The thionamide antithyroid drugs (ATDs) are used to reduce the excessive thyroid hormone synthesis and secretion. These are methimazole and carbimazole, or propylthiouracil, which inhibit TPO activity, and thus reduce the iodide organification and thyroid hormone synthesis. Beta-adrenergic blockade, with a preference for nonselective agents, assists acutely with symptom control. High-dose beta-adrenergic blocker therapy can also reduce the peripheral deiodination of T4 to T3. Management of Graves’ hyperthyroidism will often involve a prolonged 12–18-month course of ATDs to allow for immunological remission, whereas in toxic MNG, or in toxic adenoma, remission is unlikely, especially in the absence of excess iodine exposure. Hence, definitive (ablative) management with either RAI, or thyroidectomy should be considered after the ATD therapy has resulted in biochemical euthyroidism. Persistent or recurrent Graves’ hyperthyroidism is usually treated by ablative therapy. Long-term management with low-dose ATDs may be appropriate in selected cases if ablative therapy is contraindicated. RAI therapy may be used in Graves’ disease, toxic MNG, or toxic adenoma to achieve long-term euthyroidism. RAI is a less invasive treatment option, without anesthetic risk when compared with surgery. However, RAI may require more than one dose to achieve euthyroidism and carries a risk of early or late hypothyroidism, requiring surveillance to detect. RAI should be avoided in patients with active Graves’ ophthalmopathy and women of child-bearing age planning pregnancy within 6 months. Thyroidectomy may be the preferred option in the setting of women planning pregnancy in less than 6 months, large symptomatic goiters, in thyroids with insufficient uptake for RAI administration, and in the presence of large nodules with increased malignancy risk (e.g., greater than 4 cm diameter). However, it involves an invasive surgical procedure which carries both surgical and anesthetic risk, as well as requirement for life-long levothyroxine replacement [ , ].
Impact of treatment of hyperthyroidism on hypertension and cardiovascular disease
Treatment of the underlying hyperthyroidism leads to reversal of the cardiac manifestations that contribute to hypertension and may result in normalization of blood pressure without the requirement for additional antihypertensive treatment [ , ].
When to suspect hypothyroidism and hyperthyroidism in hypertensive subjects?
Endocrine causes of hypertension are important, although thyroid disorders are less prominent than pheochromocytoma, primary aldosteronism, and Cushing’s syndrome [ ]. However, disorders of thyroid function are relatively common in the general population and very easily treatable. Therefore, clinical assessment for thyroid disease and use of TSH measurement for case finding in hypertensive patients is an easy adjunct, and therefore recommended [ ].
Parathyroid disease
Physiology of parathyroid hormone action
The four parathyroid glands in human beings lie posterior to the thyroid gland in the neck. Parathyroid chief cells produce the precursor molecule preproparathyroid hormone (prepro-PTH), which is then degraded to the active 84 amino acid PTH prior to release. PTH regulates calcium and phosphate homeostasis in conjunction with calcium monitoring via the G protein-coupled calcium-sensing receptors (CaSRs). In response to a reduction in serum calcium level, PTH is secreted from the parathyroid glands and acts on the bone and the kidneys to activate the PTH cell surface receptors. PTH increases serum calcium levels via a number of distinct mechanisms. PTH directly stimulates the osteoclastic activity, and inhibits the osteoblastic activity in the skeletal system, resulting in bone breakdown and release of calcium into the blood stream. PTH directly stimulates the increased reabsorption of calcium from the distal convoluted tubules in the kidneys. Phosphate reabsorption is inhibited in the proximal and distal tubules by PTH, leading to a concurrent increase in renal phosphate excretion. PTH also stimulates the release of the active form of vitamin D 3 [1,25(OH) 2 D 3 ] from the renal tubules. 1,25(OH) 2 D 3 , in turn, stimulates the increased dietary calcium absorption from the intestines via its intracellular nuclear receptor. The increase in serum calcium and increase in 1,25(OH) 2 D 3 levels provide a negative feedback to the parathyroid glands to suppress further PTH production [ ].
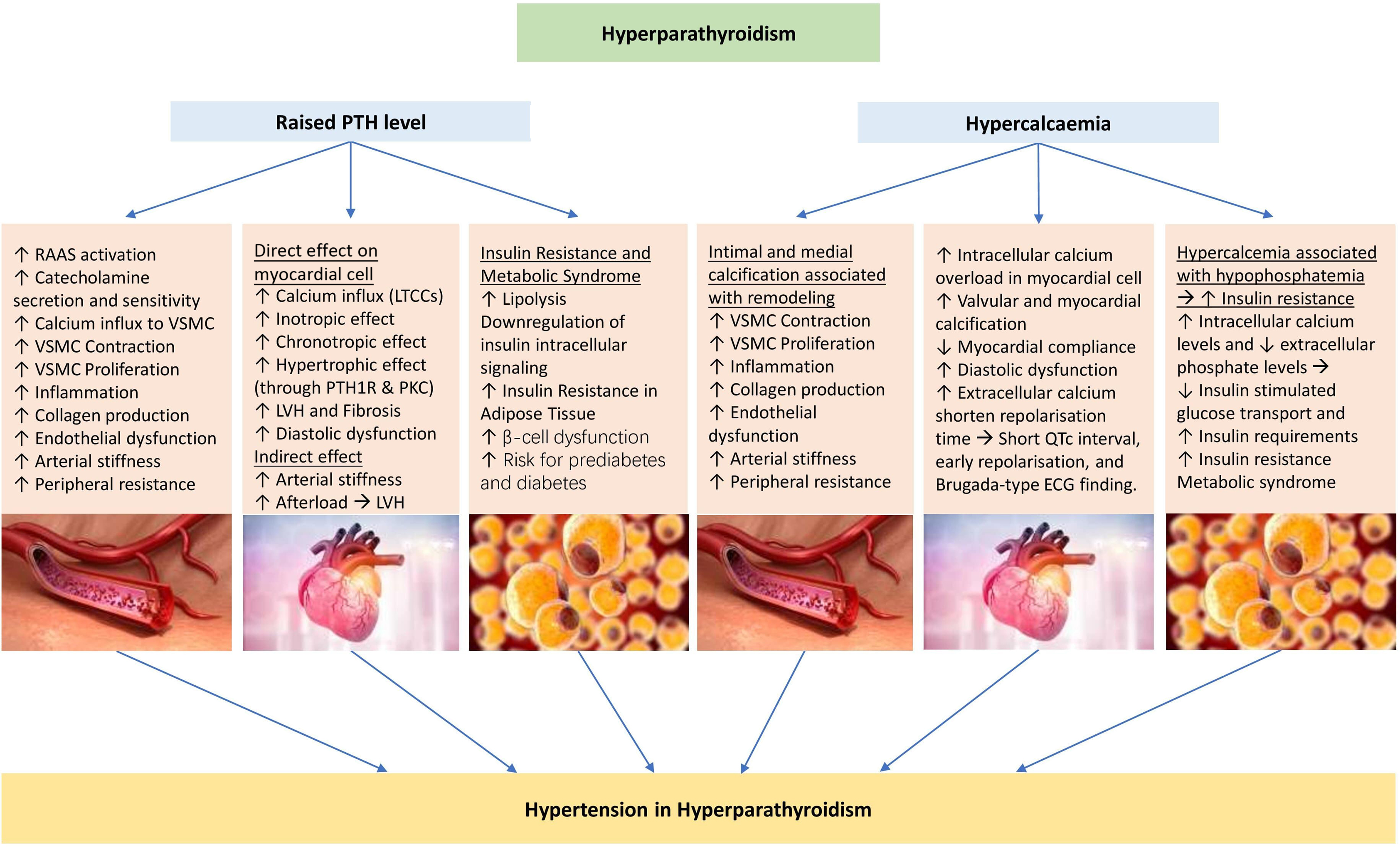
Pathophysiology of hypertension in primary hyperparathyroidism
The prevalence of hypertension in patients with primary hyperparathyroidism ranges from 40% to 65%, compared with the global prevalence of hypertension in adults of 32% and 34% in women and men, respectively [ ]. Patients with primary hyperparathyroidism also have concurrently higher rates of metabolic syndrome and its associated disorders including obesity, dyslipidemia, and diabetes mellitus. This may further contribute to the elevated rates of hypertension seen in these patients [ ]. The increased risk of metabolic syndrome has been demonstrated in patients with hypercalcemic as well as normocalcemic hyperparathyroidism, suggesting a direct relationship with PTH action, rather than the absolute serum calcium level [ ]. In fact, hyperparathyroidism associated with moderate or severe elevations in PTH may have a greater impact on calcium and bone metabolism, whereas those associated with mild elevations in PTH presenting with normocalcemia or only mild hypercalcemia are more often associated with an elevated metabolic syndrome prevalence and an increased cardiovascular disease risk [ ].
In addition to its actions on calcium homeostasis, PTH works directly on the cells of the cardiovascular system with multifactorial contributions to hypertension, though debate remains regarding the exact underlying mechanisms. Elevated serum calcium levels can lead to increased vascular calcification, driven by either endothelial or intimal dysfunction, with resultant reduced vascular reactivity, and impaired vasodilation [ ]. PTH also stimulates cyclic adenosine monophosphate (cAMP)-driven Ca 2+ influx into the vascular smooth muscle cells leading to enhanced vascular contraction, and increased peripheral vascular resistance [ , ], as well as independent markers of increased arterial stiffness [ , ]. Elevated PTH levels have also been associated with elevated secretion and sensitivity to circulating catecholamines, in particular noradrenaline, leading to further vasoconstriction [ , ]. In addition, PTH activates G protein-coupled L-type Ca 2+ channels in adult cardiac myocytes, leading to increased influx of extracellular Ca 2+ , and increased cardiac myocyte contractility [ , ]. PTH can also lead to positive chronotropic effects by activation of cAMP and cardiac L-type Ca 2+ channels leading to an influx of Ca 2+ intracellularly [ , ]. Finally, PTH can mediate stimulation of protein synthesis and proliferation of cardiac myocytes via activation of protein kinase C, resulting in ventricular hypertrophy [ , ].
Bidirectional relation between renin–angiotensin–aldosterone system and PTH
RAAS contributes to the control of blood pressure via its effects on electrolytes, fluid balance, and systemic vascular resistance. Angiotensinogen is cleaved to angiotensin I (Ang I) by the enzyme renin. Renin is released from the juxtaglomerular cells of the kidney when the afferent arteriole is stimulated by reduced renal arteriolar perfusion pressure resulting from a decreased systemic arterial perfusion (via increased circulating catecholamines). Renin is also stimulated by reduced Na + and Cl − delivery to the macula densa. The Ang I is then cleaved to form angiotensin II (Ang II) by the angiotensin-converting enzyme (ACE). Ang II activates the Ang II type 1 (AT1) receptors, which triggers vasoconstriction of the peripheral arteries and arterioles, increases sympathetic release and catecholamine sensitivity, and increases aldosterone production from the adrenal zona glomerulosa. Aldosterone increases Na + resorption from the tubular fluid via epithelial sodium channels in exchange for K + and H + ion secretion into the filtrate. Water movement follows the Na + gradient, leading to fluid retention, and expansion of the plasma volume. These multifactorial mechanisms lead to mineralocorticoid-induced hypertension [ ].
Release and activity of PTH and RAAS demonstrate a bidirectional relationship in the elevation of blood pressure. Aldosterone-secreting adenoma cells express type 1 PTH receptors and primary aldosteronism is associated with concurrent elevations in PTH levels that improve following adrenalectomy or RAAS blockade [ , ]. It is thought that the Ang II and aldosterone have a direct stimulatory effect on PTH release via the AT1 receptors that are also found on parathyroid cells [ ]. Several studies have also postulated that elevated PTH levels seen in primary aldosteronism are via an indirect mechanism due to RAAS-driven hypertension altering renal Ca 2+ handling, leading to decreased serum Ca 2+ with a resultant elevation in PTH level [ ]. An additional mechanism is the loss of the diurnal decrease in renin and aldosterone secretion that may be mediated by the hypercalcemia of primary hyperparathyroidism [ ].
Parathyroid cells also express mineralocorticoid receptors [ ], and an increase in renin secretion can also be seen in cases of primary hyperparathyroidism, which improves following parathyroidectomy [ ]. PTH activates cAMP, phospholipase C, and protein kinase-C second messenger signaling, as well as increasing intracellular Ca 2+ entry via L-type Ca 2+ channels. Intracellular Ca 2+ availability is essential for the mitochondrial matrix, a vital step for CYP11B2-driven steroidogenesis, and resultant mineralocorticoid formation [ ]. PTH-induced increase in intracellular Ca 2+ is also thought to directly stimulate aldosterone release [ ]. It appears that excess levels of both PTH and aldosterone can activate further release of each other, leading to parallel summative positive effects on blood pressure elevation.
Putative mechanisms in the development of hypertension in patients with primary hyperparathyroidism are shown in Fig. 16.3 .