Hypersensitivity Pneumonitis
Michael C. Zacharisen
Jordan N. Fink
Hypersensitivity pneumonitis, also known as extrinsic allergic alveolitis, is a multifaceted immunologically mediated pulmonary disease with associated constitutional symptoms due to sensitization and then repeated inhalation of a wide variety of inhaled organic dusts. It is characterized by nonimmunoglobulin E (IgE)-mediated inflammation of the pulmonary interstitium, terminal airways, and alveoli. This syndrome occurs in both atopic and nonatopic individuals and may present in several clinical forms depending on the duration, frequency, and intensity of antigen exposure, the antigenicity of the offending agent, and the patient’s age and immunologic responsiveness. The preponderance of cases occurs in occupational and agricultural settings. However, various hobbies and medications are also associated with hypersensitivity pneumonitis. Despite the many antigens recognized to cause hypersensitivity pneumonitis, the clinical, immunologic, and pathophysiologic findings are generally comparable.
Allergens of Hypersensitivity Pneumonitis
Hypersensitivity pneumonitis was recognized by Ramazzini in 1713 in grain workers (1). As awareness of this pulmonary disease has increased, there has been identification of new antigens implicated in the disease currently encompassing over 200 different agents (2). Although the immunopathophysiology of the disease is becoming clarified, there continue to be cases of hypersensitivity pneumonitis in which the specific antigen has not been defined. The primary exposures for the development of hypersensitivity pneumonitis are occupational, agricultural, and those related to hobbies. To reach the terminal airways and alveoli, the allergenic particles must be smaller than 3 μm to 5 μm. The variety of causative antigens includes airborne microbial antigens, animal or plant products, and low-molecular-weight chemicals (Table 23.1). Many of these same antigens, such as diisocyanates, mammalian and insect proteins, and wood dusts, also can induce IgE-mast cell mediated allergic responses including asthma.
Table 23.1 Some Antigens of Hypersensitivity Pneumonitis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Thermophilic actinomycetes were recognized as the causative agent in farmer’s lung in 1932 in England (3). These bacteria thrive at temperatures of 70°C and can be found in high concentrations in compost piles or in silos where animal fodder is stored and becomes a culture medium for the organism. Identification and clarification of the responsible antigens have been described by a number of investigators (4,5). Increased awareness of the environmental factors favoring disease and changes in farming techniques have reduced the incidence of this disorder (6).
Both commercial and residential exposures to mold-contaminated materials have been implicated in a number of cases of hypersensitivity pneumonitis with the descriptive names of many of these diseases reflecting the source of exposure. For example, ventilation pneumonitis, caused by contaminated heating or cooling units, is probably the most common building-related form of hypersensitivity pneumonitis (7,8). This syndrome may occur as a result of the inhalation of aerosols containing antigens found in small home ultrasonic humidifiers to large industrial air handling units (9). Over the past decade, respiratory illness related to inhalation of metal-working fluids containing gram-negative bacteria has been reported; this finding has far-reaching consequences for industry (10–12). While fungal exposure is ubiquitous outdoors, indoor exposure in water-damaged environments is less well characterized, but many case reports incriminate fungi as the cause of the disease in both adults and children (13). The role of fungal fragments in initiating human disease has yet to be clarified, but it provides a new paradigm for fungal exposure (14). Workers cultivating mushrooms in indoor facilities have been identified as another occupation with many affected individuals (15,16). Pigeon breeders and bird fanciers have long been recognized to develop hypersensitivity pneumonitis to inhaled antigens in dried avian droppings and feather bloom (17,18). A variety of exotic, wild, and domestic birds have also been identified as causing bird breeder’s disease (19–21).
As new cases of hypersensitivity pneumonitis are recognized, measures to identify the antigen and decrease antigen exposure can be implemented. This recognition, as well as changes in exposure, has resulted in some hypersensitivity diseases such as smallpox handler’s lung and pituitary snuff taker’s lung (porcine and bovine allergens) being of historical interest only (22). Occupational exposures recently recognized include the manufacture of yacht hulls where inhalation of fumes from heated chemicals in rolling fiberglass has been implicated (23). Kiln-dried wood heavily contaminated with Paecilomyces has affected workers in a hardwood
floor processing plant (24). Inhalation of the coolant HFC134a used during laser removal of body hair has been reported to trigger HP symptoms with peripheral blood and bronchial biopsy eosinophilia (25).
floor processing plant (24). Inhalation of the coolant HFC134a used during laser removal of body hair has been reported to trigger HP symptoms with peripheral blood and bronchial biopsy eosinophilia (25).
Medications are also an important cause of pulmonary disease that resembles hypersensitivity pneumonitis. Among the implicated medications are nitrofurantoin, amiodarone, minocycline, roxithromycin, lenalidomide, nadolol, and sulphasalazine (26–31). Intranasal heroin has also been reported to cause the syndrome (32).
Specific syndromes of hypersensitivity pneumonitis occur in different parts of the world. For example, esparto grass is used in the production of rope, matting, paper pulp, and plaster in Mediterranean countries. Individuals such as stucco workers have developed hypersensitivity pneumonitis to Aspergillus fumigates-contaminated esparto fiber dust in their workplace environments (33). Workers in Eastern Canada who are employed in peat moss processing plants are frequently exposed to loose dry material which may contain many microorganisms, of which molds have been implicated in causing hypersensitivity pneumonitis (34). Summer-type hypersensitivity pneumonitis due to Trichosporon is an important example of a disease not found in the United States, but is the most prevalent form of hypersensitivity pneumonitis in Japan (35). In the Midwest United States, of 85 patients with hypersensitivity pneumonitis identified from 1997 to 2002, the most common causes were avian-related (34%), hot tub lung (21%), farmer’s lung (11%), household mold exposure (9%), and unidentified antigen (25%) (36). Controversy surrounds the classification of “hot tub lung” as hypersensitivity pneumonitis versus infection with nontuberculous mycobacteria.
Epidemiology
The exact incidence of hypersensitivity pneumonitis is unknown, but it has been identified in 2% to 8% of farmers (37) and in 6% to 21% of pigeon breeders (38). Of 36 cases of chronic hypersensitivity pneumonitis identified by a hospital survey in Japan, reported etiologies were summer-type hypersensitivity pneumonitis (10 cases), other home-related causes (5 cases), bird fancier’s disease (7 cases), isocyanate (5 cases), farmer’s lung (4 cases), and five miscellaneous cases (39). In Ireland, haymaking methods were revolutionized in the 1980s and between 1997 and 2002, a marked decline in hypersensitivity pneumonitis was observed (40). In the United Kingdom, however, the overall incidence of hypersensitivity pneumonitis is stable at 600 new cases per year (1 per 100,000 general population) (41). In the United States, the “healthy worker effect” and high employee turnover may be partly responsible for the underreporting or underrecognition of work-related cases of hypersensitivity pneumonitis.
Diagnostic Criteria and Clinical Features
The criteria for the diagnosis of hypersensitivity pneumonitis consist of recognizing the clinical features with supporting exposure history, laboratory, pulmonary function, and radiographic characteristics (Fig. 23.1.) (42). While there is no single confirmatory test for hypersensitivity pneumonitis, not even lung biopsy, six significant predictors were identified that provide a 95% confidence interval. These include: (a) exposure to a known offending allergen; (b) positive precipitating antibodies to the offending antigen; (c) recurrent episodes of symptoms; (d) inspiratory crackles on lung auscultation; (e) symptoms occurring 4 to 8 hours after exposure, and (f) weight loss (43). The clinical presentation follows repeated exposure and can vary from sudden and explosive systemic and respiratory symptoms to an insidious, progressive course of dyspnea, fatigue, and weight loss. Based on these clinical presentations, hypersensitivity pneumonitis has been divided into acute, subacute, and chronic forms (44).
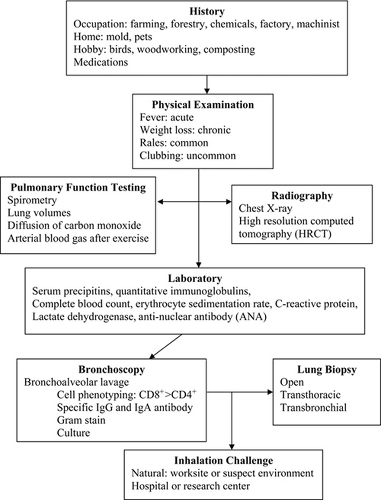 Figure 23.1 Evaluation of hypersensitivity pneumonitis. |
The patient with the acute form presents with nonproductive cough, dyspnea, sweating, myalgia, and malaise occurring 4 to 12 hours after intense exposure to the inciting allergen. Acute viral or bacterial infections may mimic this presentation, leading to treatment with antibiotics. With avoidance of the allergen, the symptoms spontaneously resolve over 18 to 24 hours, with complete resolution within days. This is in contrast to viral infections. On repeat exposure, the symptoms recur with either more severe and progressive symptoms or less intense and nonprogressive symptoms. The patient may recognize this pattern and try to minimize their exposure. The chronic form is characterized by the insidious onset of dyspnea that especially occurs with exertion. Other symptoms include productive cough, fatigue, and anorexia with weight loss. Fever is not typical unless there is a high-dose allergen exposure superimposed on the chronic symptoms. This form is usually related to continuous low-level antigen exposure and is not often recognized resulting in a delay in the correct diagnosis. An antigen exposure history could be the only clue to the diagnosis. The subacute form is characterized by symptoms intermediate to the acute and chronic form with progressive lower respiratory symptoms. The acute and subacute forms may overlap clinically, just as the subacute and chronic forms may.
Physical Examination
The physical examination may be normal in the asymptomatic patient between widely spaced episodes of acute hypersensitivity pneumonitis. Fine, dry rales may be present, depending on the degree of lung disease present and the timing following the most recent exposure. Wheezing is not a prominent symptom. An
acute flare-up of hypersensitivity pneumonitis is associated with an ill-appearing patient in respiratory distress with temperature elevation up to 40°C 6 to 12 hours after antigen exposure. Rash, lymphadenopathy, or rhinitis should prompt investigation for causes other than hypersensitivity pneumonitis. With extensive fibrosis that occurs in the chronic form of the disease, dry rales and decreased breath sounds predominate. Some patients with end-stage disease may have digital clubbing (45).
acute flare-up of hypersensitivity pneumonitis is associated with an ill-appearing patient in respiratory distress with temperature elevation up to 40°C 6 to 12 hours after antigen exposure. Rash, lymphadenopathy, or rhinitis should prompt investigation for causes other than hypersensitivity pneumonitis. With extensive fibrosis that occurs in the chronic form of the disease, dry rales and decreased breath sounds predominate. Some patients with end-stage disease may have digital clubbing (45).
Pulmonary Function Tests
The classic pulmonary function abnormality in the acute form is restriction with decreased forced vital capacity (FVC) and forced expiratory volume in 1 second (FEV1) occurring 6 to 12 hours after exposure to the offending antigen ( Fig. 23.2). A biphasic obstructive response similar to that seen in the early and late phases of asthma has been observed in patients who develop both asthma and hypersensitivity pneumonitis as a result of sensitization to the same antigen. Peripheral airways obstruction as determined by decreased FEV1 and/or forced midrange flow measurements (FEF25-75%) has frequently been reported. Decreased gas transfer across the alveolar wall as measured by the diffusion capacity of carbon monoxide (DLCO) is often detected. This is in contrast to asthma, a disease in which an elevated DLCO commonly occurs. Although hypoxemia at rest may be observed with severe lung damage, hypoxemia with exercise is common and can be documented by pre- and postexercise arterial blood gas measurements. Bronchial hyperresponsiveness as determined by methacholine challenge is present in a majority of patients with hypersensitivity pneumonitis and is likely due to the inflammatory response of the airways. In subacute and chronic hypersensitivity pneumonitis, there is usually a demonstrable combination of obstruction and restriction.
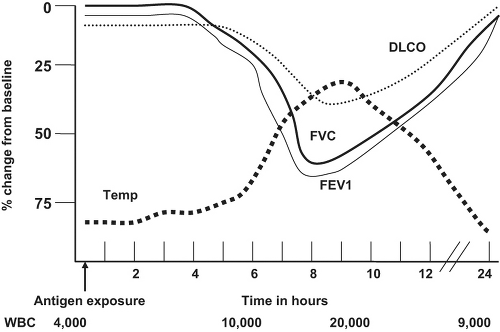 Figure 23.2 Graphic representation of changes in acute hypersensitivity pneumonitis. |
Radiographic Features
Chest Radiographs
Radiographic abnormalities may be transient or permanent depending on the form or stage of disease. Transient radiographic changes occur primarily in the acute form with patchy, peripheral, bilateral interstitial infiltrates with a fine, reticulonodular pattern similar to acute pulmonary edema (46) as seen in Figure 23.3. There may be bilateral ground-glass opacities in the middle to lower lung fields that are indistinguishable from other interstitial lung disorders. Central lymphadenopathy also may be present. These changes usually resolve spontaneously with avoidance or with corticosteroid therapy. Between acute attacks, the chest radiograph is usually normal.
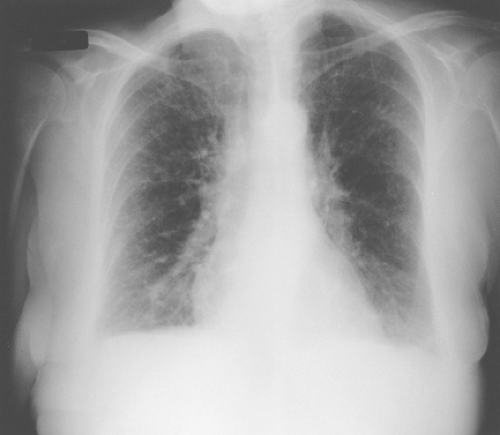 Figure 23.3 Chest radiograph of a patient with hypersensitivity pneumonitis demonstrating bilateral lower lobe patchy infiltrates and a reticulonodular pattern. |
In the subacute form, nodular, patchy, or diffuse infiltrates with bilateral ground-glass opacities; poorly defined small centrilobular nodules; and lobular areas of decreased attenuation, vascularity on inspiration, and of air-trapping on expiration have been observed (47).
In the chronic form, fibrotic changes with patchy or random reticulation, traction bronchiectasis, and areas of emphysema may be seen superimposed on acute or subacute changes, typically sparing the lung bases. Less commonly, subpleural honeycombing is found (47). Findings not characteristic of hypersensitivity pneumonitis include calcification, cavitation, atelectasis, solitary pulmonary nodules, pneumothorax, and pleural effusions.
Computed Tomography Scans
Related posts:
 Immunology of IgE-mediated and Other Hypersensitivity Responses
Immunology of IgE-mediated and Other Hypersensitivity Responses
 Physiologic and Biologic Evaluation of Allergic Lung Diseases
Physiologic and Biologic Evaluation of Allergic Lung Diseases
 Erythema Multiforme, Stevens-Johnson Syndrome, and Toxic Epidermal Necrolysis
Erythema Multiforme, Stevens-Johnson Syndrome, and Toxic Epidermal Necrolysis
 Management of Acute Severe Asthma
Management of Acute Severe Asthma
 Nasal Polyposis, Sinusitis, and Nonallergic Rhinitis
Nasal Polyposis, Sinusitis, and Nonallergic Rhinitis
 Eosinophilic Esophagitis
Eosinophilic Esophagitis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


