Hydroxyurea (HU), one of the simplest of the anticancer drugs, plays a supportive role in the treatment of myeloproliferative disease because of its ability to suppress proliferation of myeloid, erythroid, and platelet precursors, but its value is limited by its inability to induce bone marrow remission and the equally rapid reversibility of its myelosuppressive effect. However, it has other notable clinical properties, including its induction of
β-globin synthesis in patients with sickle cell anemia and thalassemia. It has been an invaluable probe for the laboratory study of its intracellular target, ribonucleotide reductase (RR), the rate-limiting step in the de novo synthesis of deoxyribonucleotide triphosphates (dNTPs). Other actions, including the generation of nitroxyl radicals and radiosensitizing effects, have potential clinical applications. The key features of this drug are shown in
Table 12-1.
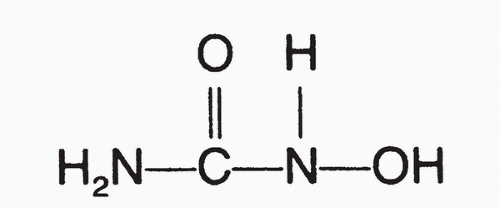
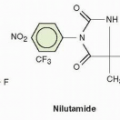
HU (
Fig. 12-1) was originally synthesized in Germany in 1860,
1 and it was found to have inhibitory effects on granulocyte production.
2 It displayed antileukemic properties in the National Cancer Institute’s screening system,
3 entered clinical trials in the 1960s, and was soon recognized as a potent myelosuppressive agent with a novel mechanism of action and few side effects, properties that have earned it a limited but constant role in cancer chemotherapy. Other inhibitors of RR have since been evaluated in the clinic, including compounds of the thiosemicarbazone series
4 and guanazole,
5 but they have no special therapeutic advantage and greater toxicity. The principal use of HU at present is in controlling lineage proliferation; it provides excellent control of myeloproliferative disorders.
6,
7,
8Once a primary agent with interferon-α in first-line therapy against chronic myelogenous leukemia (CML),
8 it has been largely replaced by the targeted agent, imatinib mesylate, and is now used primarily for acute control of white cell count at presentation or during blastic transformation. In polycythemia vera (PV), it effectively prevents thrombosis resulting from elevated hematocrit and high platelet count,
6 and it similarly lessens the incidence of thrombosis in patients with essential thrombocythemia (ET) and platelet counts above 1.5 million.
7 Because both PV and ET are chronic, slowly progressive diseases, there is concern that HU may increase the risk of leukemic conversion, a risk that has not been substantiated thus far.
9,
10 In younger patients with PV, who have the prospect of long-term treatment, prophylactic phlebotomy is the favored treatment, and in patients with ET, anagrelide and interferon-α are alternatives to HU. Newer drugs that attack the mutation in Jak-2 kinase have entered clinical trial for PV and ET and may displace HU in the future.
A major current use of HU is in the prevention of complications of sickle cell anemia. In patients with sickle cell anemia, HU increases the production of fetal hemoglobin, ameliorates symptoms, and reduces the incidence of painful crisis and hospitalization.
11,
12 In vitro incubation of HU with erythroid progenitors induces fetal hemoglobin
β-globin production.
13 Whether the induction of fetal hemoglobin represents a response to inhibition of DNA synthesis in red cell progenitors or a specific alteration of
γ-globin gene transcription is uncertain.
13 Nitroxy radicals produced by decomposition of HU may directly stimulate
γ-globin gene transcription through the Sar 1a promoter.
14,
15 The increase in fetal hemoglobin promotes the solubility of hemoglobin and prevents the downward spiral of intravascular red cell sickling that leads to painful crisis. Convincing evidence now exists that induction of fetal hemoglobin is not the only, and perhaps not the major, contributor to the drug’s efficacy. The benefit from HU may be partly related to its ability to suppress the neutrophil count and its effects on white and red cell adhesion to vessel walls.
16 A marked decrease in the endothelial adhesion of a patient’s blood cells, coincident with a down-regulation of L-selectin on the red cell and white cell surfaces, is observed after 2 weeks of HU therapy, before fetal hemoglobin levels rise. There is a strong inverse correlation between neutrophil count and crisis rate. In terms of clinical efficacy, a randomized, double-blind study has demonstrated that long-term treatment with HU decreases the incidence of painful crisis by 44% in adult patients with sickle cell disease.
17 HU treatment the frequency of acute chest syndrome and hospitalization and the need for blood transfusion. These results establish HU as the first clinically acceptable drug shown to decrease crises in sickle cell disease. HU appears to be as effective in children with sickle cell disease
18 and in patients with sickle cell-
β-thalassemia and sickle cell-hemoglobin C disease.
19
Mechanism of Action and Cellular Pharmacology
The primary site of cytotoxic action for HU is inhibition of the RR enzyme system. This highly regulated enzyme system is responsible for the conversion of ribonucleotide diphosphates to the deoxyribonucleotide form, which can subsequently be used in either de novo DNA synthesis or DNA repair.
20 HU inhibits RR in vitro,
21 and the extent of inhibition of DNA synthesis observed in HU-treated cells correlates closely with the size of the decreased deoxyribonucleotide pools.
22 This enzyme has an important role as a rate-limiting reaction in the regulation of DNA synthesis. In human and other mammalian cells, this unique enzyme consists of two different subunits, usually referred to as M-1 and M-2.
23 Protein M-1 is a dimer with a molecular weight of 170 kD and contains the binding site
for the diphosphate substrates as well as the allosteric nucleotide triphosphate regulatory sites.
24 Although considerable variability exists among enzymes from various tissue sources, the general regulatory effects are summarized in
Table 12-2. The reduction of all substrates is inhibited and the enzyme complex dissociates in the presence of deoxyadenosine triphosphate.
25 Protein M-1 is present at a relatively constant level throughout the cell cycle, except in cells in G
0 or those that have undergone terminal differentiation, in which it is markedly decreased.
26 The gene coding for the M-1 protein can be mapped to chromosome 11.
26Protein M-2 is the catalytic subunit of the enzyme and exists as a dimer with a molecular weight of 88 kD. This unique protein contains stoichiometric amounts of iron and a stable organic free radical localized to a tyrosine residue. The fully conserved tyrosyl radical is essential to enzyme activity and is localized in proximity to and stabilized by the binuclear nonheme iron complex.
27 The cellular concentration of M-2 protein is variable throughout the cell cycle; it peaks in S phase, which suggests that functional enzyme activity depends on the concentration of M-2 protein.
28 The M-2 subunit sequences have been mapped to chromosome 2 in human cells and seem to be in the same amplification unit as the gene for ornithine decarboxylase.
HU enters cells by passive diffusion. The inhibition of RR occurs as a result of the drug’s chelation of iron and its inactivation of the tyrosyl free radical on the M-2 subunit, with disruption of the enzyme’s iron-binding center.
29 The fact that this inhibition can be partially reversed in vitro by ferrous iron and that cytotoxicity can be enhanced by iron-chelating agents
30 emphasizes the importance
of the nonheme iron cofactor in this process. HU selectively kills cells in S phase, and within an S-phase population of cells, those that are most rapidly synthesizing DNA are most sensitive.
31 The cytotoxic effects of HU correlate with dose or concentration achieved, as well as with duration of drug exposure.
32 Following HU exposure, cells progress normally through the cell cycle until they reach the G
1-S interface. Rather than being prevented from entering S phase, as was once thought, cells enter S phase at a normal rate but are accumulated there as a result of the inhibition of DNA synthesis.
33 Cells undergo apoptosis in a process mediated by both
p53 and non-
p53 pathways.