Summary of Key Points
- •
Humoral and cellular immune dysregulation in the tumor microenvironment contributes to immune evasion, a key hallmark of lung cancer.
- •
Immunosuppressive mechanisms observed in lung cancer include defective antigen presentation, secretion of immunosuppressive tumor-derived soluble factors, and immunosuppressive cells infiltrating the tumors.
- •
Suppression of antigen-presenting machinery in lung cancer results from several mechanisms including deficiencies in expression of antigen-processing genes and haplotype loss of human leukocyte antigen (HLA) class I antigens.
- •
Immune inhibitory cytokines secreted by the tumor cells impair T-cell survival and help avoid T cell-mediated immune responses.
- •
Immune checkpoints expressed on the surface of T lymphocytes modulate the immune response to antigens via inhibitory or stimulatory signaling to T cells.
- •
Tobacco smoke markedly influences the immune microenvironment in lung cancer.
Advances in the understanding of cellular immunology and tumor–host immune interactions have led to the development of promising immunotherapies in lung cancer. This chapter provides a review of the current understanding of the basic immunologic abnormalities in lung cancer. Clinical trials of immunotherapy in lung cancer are discussed in Chapter 50 .
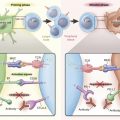
Although lung cancer was traditionally thought to be a nonimmunogenic tumor unlike melanoma or renal cell cancer, accumulating evidence suggests both cellular (T lymphocyte-mediated) and humoral (antibody-mediated) immune antitumor responses even in patients with advanced lung cancer. Despite the immune responses, spontaneous tumor regressions rarely occur, indicating the ability of the tumor cells to escape an immune response. In fact, lung cancer is among the many tumors that are known to promote immune tolerance and escape host immune surveillance. It is thought that the immune system actively inhibits the formation and progression of transformed cells and ultimately “shapes” nascent tumors by forcing the selective evolution of tumor cells that can evade the immune response, a phenomenon called tumor immunoediting. Tumors also utilize numerous other pathways to inhibit immune responses, including local immune suppression, induction of tolerance, and systemic dysfunction in T-cell signaling. Although these immunosuppressive mechanisms are categorized discretely, the clinically observed deficits are interrelated ( Fig. 16.1 ).
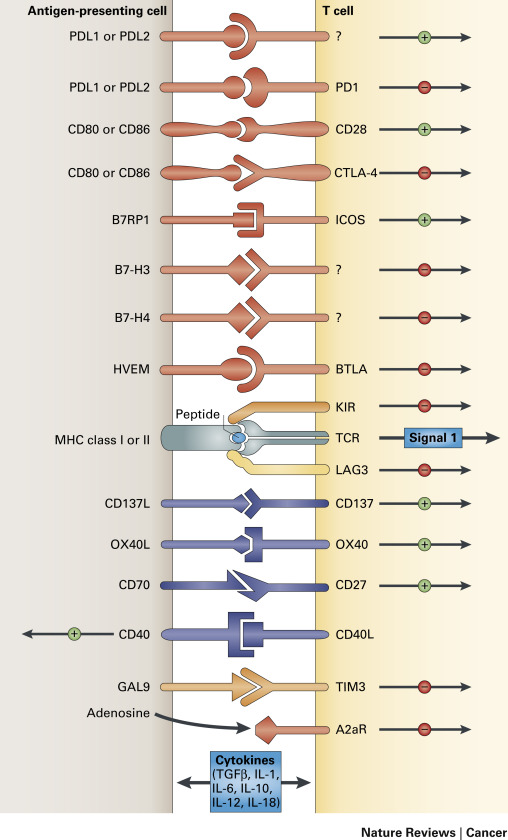
Suppression of the Antigen-Presenting Machinery
An adaptive immune response requires two signals between the antigen-presenting cells (APCs) and the effector T cell. The first signal is mediated by the T-cell receptor and the specific antigenic peptide presented in the context of major histocompatibility complex (MHC) class I or class II molecules expressed on the APC surface. The second signal is mediated through constitutively expressed costimulatory molecules on the T cell (CD28) and the APC (B7-1/CD80 or B7-2/CD86; Fig. 16.1 ). The presence of both signals trigger intracellular events resulting in the activation and interleukin-2 (IL-2)-dependent clonal proliferation of T cells.
The MHC class I molecules are essential components of the adaptive immune system and are crucial to the immune recognition of tumor cells. MHC class I molecules report on cellular transformation to CD8 + cytotoxic T lymphocytes (CTLs) through a multistep process of antigenic peptide acquisition, tagging them for destruction by ubiquitylation, proteolysis, delivery of peptides from cytosol to the endoplasmic reticulum via the heterodimeric transporter associated with antigen processing (TAP) 1 and TAP2 subunits, binding of peptides to MHC class I molecules, and displaying of peptide–MHC class I complexes on the cell surface.
Under physiologic conditions, the components of MHC class I antigen-processing machinery (APM) are constitutively expressed in all adult nucleated cells (except immune privileged tissue). Their expression is regulated by cytokines that can alter the surface expression of MHC class I molecules. Aberrant MHC class I expression has been conclusively demonstrated as an important immune escape mechanism in cancers. MHC class I abnormalities have been frequently found in a variety of human cancers, are associated with unfavorable prognoses in some tumor types, and have a negative impact on the outcome of T cell-based immunotherapy. Marked deficiency or lack of expression of MHC class I molecules has been demonstrated in lung cancer.
The molecular mechanisms of MHC class I expression loss are diverse and include structural alterations or dysregulations of genes encoding the classical MHC class I antigens and/or components of the MHC class I APM. The dysregulation of APM components may occur at the epigenetic, transcriptional, or posttranscriptional level. Mechanisms underlying the aberrant expression of MHC class I antigens in lung cancer include deficiencies in the expression of antigen-processing genes (e.g., genes that encode proteasome subunits and the peptide transporters), which result in defective peptide transport to the cell surface from the endoplasmic reticulum. Haplotype loss of HLA class I antigens is another mechanism of abnormal HLA expression in lung cancer and has been demonstrated in about 40% of lung cancer cell lines. Structural alterations such as β2-microglobulin gene abnormalities resulting from loss of messenger RNA and point mutations are less common mechanisms of altered MHC class I expression in lung cancer.
Studies in which lung cancer cell lines with haplotype loss of HLA class I antigens are used indicate that tumor cells with a normal HLA class I expression may be killed by CTLs at an early stage of carcinogenesis, and only immunoselected tumor cells that lack HLA class I expression can escape this immune attack and develop into cancer. Furthermore, some defects in MHC class I expression in lung cancer, for example, deficiencies in expression of antigen-processing genes and not β2-microglobulin gene abnormalities, may be reversible with cytokines. Interferon-γ ( IFN-γ ) gene transfection into HLA-deficient small cell lung cancer (SCLC) was able to restore its ability to present endogenous tumor antigens to CTL with a concomitant increase in cell-surface expression of class I molecules.
The results of immunohistochemical studies of surgically resected samples with monoclonal antibodies against a common framework determinant of HLA class I antigens have shown deficient expression in HLA class I antigens in 25% to 33% of nonsmall cell lung cancer (NSCLC). As evident from studies of transfected tumor cells lacking expression of these antigens, MHC class I molecules are required for presentation of antigens on tumor cells to the CTL. Thus tumor cells that have lost MHC class I antigens have the advantage of being able to escape lysis by CTL. In lung cancers, the absence of expression of HLA class I molecules has been associated with poor differentiation and aneuploidy, suggesting that lung cancers with abnormal HLA expression may be biologically more aggressive. Taken together, these findings may suggest that tumors with aberrant expression of HLA class I molecules are associated with a poorer prognosis. However, the prognostic implications of HLA class I antigen downregulation in NSCLC are not clear.
Despite expressing antigens recognizable by the host immune system, tumors are very poor at initiating effective immune responses. However, antigen presentation alone is insufficient to activate T cells. In addition to T-cell receptor engagement of an antigenic peptide bound to MHC molecules, additional costimulatory signals are necessary for T-cell activation. The most important of these costimulatory signals is provided by the interaction of CD28 on T cells with its primary ligands B7-1 (CD80) and B7-2 (CD86) on the surface of APCs.
Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), a member of the immunoglobulin super family and homolog of CD28, binds members of the B7 family with a much higher affinity than CD28. In effect, CTLA-4 competes with CD28 for binding to the B7 family. Upregulation of surface CTLA-4 follows clonal expansion of T cells and regulates the immune response by inducing inhibitory signals in effector T cells that lead to the dampening of the effector T-cell response. CTLA-4 is thus one of the endogenous immune checkpoints that normally terminate immune responses after antigen activation. These T cells are eventually eliminated via apoptosis. CTLA-4 may also contribute to the immune-suppressive function of T-regulatory cells. Upregulation of CTLA-4 on the surface of T-regulatory cells results in suppression of activation and expansion of effector cells specific for both normal self-antigens and tumor antigens. CTLA-4 is constitutively expressed in NSCLC cell lines, where it induces apoptotic cell death on engagement with soluble B7-1 and B7-2 recombinant ligands. Furthermore, CTLA-4 expression in resected early-stage NSCLC was shown to be a good prognostic indicator in a retrospective analysis that was limited by a small number of patients.
Programmed cell death 1 (PD-1) is another key immune checkpoint receptor that is structurally similar to CTLA-4, but with distinct biologic functions and ligand specificity that is expressed by activated T cells and mediates immunosuppression. PD-1 functions primarily in peripheral tissues, where T cells may encounter the immunosuppressive PD-1 ligands PD-L1 (B7-H1) and PD-L2 (B7-DC), which are expressed by tumor cells, stromal cells, or both ( Fig. 16.2 ). The immune inhibitory signals mediated by CTLA-4 and PD-1 are different, as evidenced by the early mortality of CTLA-4 knockout mice compared with modest late-onset strain-specific and organ-specific autoimmunity of PD-1 knockout mice.
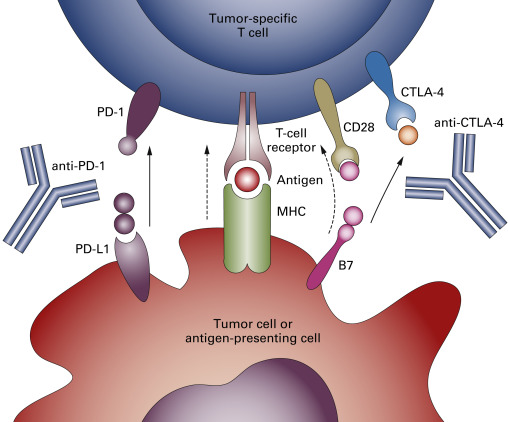
Most lung cancer samples, but not samples with normal alveolar cells, express high levels of PD-L1, which is limited to the tumor plasma membrane or cytoplasm. Dendritic cells (DCs) isolated from tumoral and nontumoral lung tissues express low, although significantly higher, levels of B7-1 and B7-2 molecules compared with blood DCs. In surgically resected NSCLC, no relationship was found between the expression of PD-L1 or PD-L2 and clinicopathologic variables, such as histology, stage, or postoperative survival. In a limited subset of patients, fewer tumor-infiltrating lymphocytes (TILs) were identified in PD-L1-positive tumor regions than in PD-L1-negative tumor regions. Tumor-infiltrating and circulating CD8 + T cells from patients with NSCLC showed increased PD-1 expression compared with peripheral blood mononuclear cells from normal volunteers, but showed impaired immune function, including reduced cytokine production capability and impaired capacity to proliferate. Blocking the PD-1 and PD-L1 pathway by anti-PD-L1 antibodies increased cytokine production and proliferation of PD-1 + tumor-infiltrating CD8 + T cells.
Tumor-Derived Soluble Factors
Tumor cells avoid lymphocyte-mediated immune responses by secreting immune inhibitory cytokines in the tumor milieu. In addition to secreting immune-suppressive mediators, tumor cells may also signal surrounding inflammatory cells to release immune-suppressive mediators, augment the trafficking of suppressor cells to the tumor site, and promote the differentiation of effector lymphocytes to a T-regulatory phenotype. Tumor-derived soluble factors also impair T-cell survival. NSCLC cell line supernatants contribute to enhanced activation-induced T-cell apoptosis in the tumor environment. The increased T-cell apoptosis after mitogen stimulation is due to the inhibition of nuclear factor-κB activation in the tumor milieu.
Interleukin-10
Human bronchial epithelial cells constitutively produce IL-10, which regulates the local immune response in normal lungs. Although the precise role of IL-10 in carcinogenesis remains controversial, results of several studies suggest that IL-10 is a potent immunosuppressive molecule that may promote lung cancer growth by suppressing T-cell and macrophage function and enabling tumors to escape immune detection. In vitro, human lung tumors produce greater amounts of IL-10 than normal lung tissue. Tumor cells also induce T lymphocyte-derived IL-10 via a prostaglandin E 2 (PGE 2 )-mediated pathway. In vivo, lung carcinoma cells grow more rapidly in IL-10 transgenic mice compared with controls. IL-10 transgenic mice also demonstrate a reduced capacity for antigen presentation, CTL generation, and type 1 cytokine production, reflecting defects in both T-cell and APC function. Increased IL-10 expression of tumor-associated macrophages in patients with NSCLC correlates with late-stage disease and other adverse prognostic features, suggesting its potential role in the progression of NSCLC. Furthermore, in patients with advanced-stage NSCLC receiving platinum-based chemotherapy, an elevated baseline serum IL-10 level was an independent predictor of poorer survival. In patients with early-stage NSCLC, an elevated IL-10 messenger RNA level was associated with worse survival.
However, preclinical and clinical models suggest that IL-10 possesses an immunostimulating property that favors immune-mediated rejection of cancer. In patients with NSCLC, the percentage of CD8 + cells expressing IL-10 was higher in cancerous tissue than in cancer-free tissue and correlated with both early-stage tumors and better survival rates. In clinical studies, lack of tumor IL-10 expression was associated with a worse disease-specific survival rate in patients with early-stage NSCLC.
Conflicting evidence on the immune regulatory function of IL-10 likely reflects its pleiotropic ability to positively or negatively influence the function of innate and adaptive immunity, resulting in immunostimulation or immunosuppression depending on its source (tumor compared with tumor-infiltrating immune cells) and on the interactions with factors found in the tumor microenvironment.
Transforming Growth Factor-β
Transforming growth factor-β (TGF-β) is a member of a family of multifunctional proteins that through complex cell signaling pathways regulate cell proliferation, differentiation, and angiogenesis. Three highly conserved and tissue-specific TGF-β isoforms, namely, TGF-β1 , TGF-β2 , and TGF-β3 , signal through heteromeric complexes of three cell-surface receptors, that is, the type III TGF-β receptors: TβRIII, TβRII, and TβRI. TGF-β ligands initiate signaling by binding to and bringing together TβRI and TβRII receptor serine and threonine kinases on the cell surface. This process allows TβRII to phosphorylate the TβRI kinase domain, which propagates the signal through phosphorylation of the small mothers against decapentaplegic (SMAD) proteins, which then translocate into the nucleus and interact in a cell-specific manner with transcription factors to regulate specifically the transcription of a multitude of TGF-β -responsive genes. While TGF-β acts as a tumor suppressor via its effects on proliferation, replication potential, and apoptosis, it also acts as a tumor promoter via its effects on migration, invasion, angiogenesis, and the immune system. TGF-β enables tumors to evade immune surveillance and kill through various mechanisms, most of which converge on the impairment of tumor cell killing by immune effector cells.
Normal bronchial epithelial cells have high-affinity receptors for TGF-β. TGF-β inhibits proliferation and induces differentiation of normal bronchial epithelial cells. TGF-β secreted by tumor cells mediates conversion of CD4 + CD25 − T cells to regulatory T (T-reg) cells. Neutralization of TGF-β abrogates this conversion both in vitro and in vivo. Cigarette smoking promotes tumorigenicity partly by abrogating TGF-β -mediated growth inhibition and apoptosis by reducing expression of SMAD3. Both SCLC and NSCLC overexpress TGF-β, and high levels of TGF-β are also detected in the serum of patients with lung cancer compared with normal individuals. Elevated plasma levels of TGF-β confer a poorer prognosis for patients with lung cancer.
A variety of other tumor-derived soluble factors contribute to the immunosuppressive milieu including vascular endothelial growth factor, PGE 2 , soluble phosphatidylserine, soluble Fas, soluble Fas-L, and soluble MHC class I–related chain A proteins. Although deposited at the primary tumor site, these factors can extend immunosuppressive effects to local lymph nodes and the spleen, thereby promoting invasion and metastasis.
Tumor-Infiltrating T Lymphocytes
The lymphocyte population that infiltrates the tumor is quite heterogeneous and consists of many different clones of lymphocytes that contain a variety of cell surface markers. The protumor and antitumor effects of interactions between tumor and immune cells in the tumor microenvironment are influenced not just by the type of immune cells (CD8 + , CD4 + , CD20 + , and forkhead box P3 [FoxP3 + ]), but also by their density and location within the tumors. In NSCLC, tumor stroma inflammatory cells are mainly lymphocytes (approximately two-thirds, and among them 20% B cells and 80% T cells) and tumor-associated macrophages (approximately one-third), with a low percentage of DCs and a very low percentage of natural killer (NK) cells. Although the cell surface markers and distribution among various lymphocyte subtypes are similar, TILs are functionally different from lymphocytes that are found in regional lymph nodes and peripheral blood. TILs are markedly suppressed in their functional capacity based on assessments of their proliferative and cytotoxic activities. In addition, tumor-infiltrating NK cells have markedly diminished activity compared with peripheral blood lymphocytes.
Using genetically engineered mouse lung adenocarcinoma into which exogenous antigens were introduced to mimic tumor neoantigens, DuPage et al. showed that endogenous T cells responded to and infiltrated tumors early during tumor development, substantially delaying malignant progression. However, despite continued antigen expression, T-cell infiltration did not persist and tumors ultimately escaped immune attack. Moreover, very few tumor-reactive T cells in the lungs were functional, as evidenced by their limited capacity to produce both IFN-γ and tumor necrosis factor-alpha (TNF-α). Immunohistochemical analysis in patients with NSCLC demonstrated significantly higher CD8 + T-cell counts within the tumor when compared with the invasive margin. However, the number of peritumoral CD8 + T cells correlated with the IFN-γ production, whereas this association was not observed intratumorally, suggesting that CD8 + T cells are able to infiltrate the tumor, but they are not able to mount a robust antitumor response once within the tumor nest.
Although TILs are associated with a favorable prognosis in many malignancies, the association between CD8 + T-cell infiltration and prognosis is controversial in lung cancer. However, high numbers of CD4 + T cells within cancer cell nests positively correlated with a favorable prognosis, suggesting that CD4 + T cells may be required for initiating and maintaining antitumor immune responses, as without CD4 + T-cell help, the resultant CD4-unhelped CD8 + T cells do not differentiate into sustainable memory cells.
Although TILs accumulate in lung cancer tissues, they fail to mount an immune response to tumor cells, in part because high proportions of NSCLC TILs are T-reg cells. TILs that are CD4 + CD25 + , the activated phenotype of T-reg cells, inhibit T-cell proliferation and prevent the host from mounting an immune response to tumor antigens. A preponderance of these T-reg cells could lead to a failure of tumor immunosurveillance or to enhanced tumor growth. In patients with NSCLC, T cells derived from the tumor show considerably increased proportions of CD4 + CD25 + T cells that produce TGF-β. These cells uniformly express high levels of CTLA-4 on their cell surface. In addition, CD4 + CD25 + T cells isolated from tumors mediate potent inhibition of the proliferation of autologous peripheral blood T cells stimulated by anti-CD3 or anti-CD3 and anti-CD28. An increased pool of CD4 + CD25 + T-reg cells with potent immunosuppressive features also exists in the peripheral blood of patients with NSCLC.
Although the activation of T-reg cells is considered antigen specific, their immunosuppressive function is nonspecific and results from inhibition of multiple phases and events in the immune response from antigen presentation to effector functions. Secretion of immunosuppressive cytokines such as TGF-β may play a role in the immune-suppressive function of T-reg cells. However, TGF-β may not be required for inhibition of proliferation of T-reg cells in patients with lung cancer: TGF-β neutralization did not abrogate the suppressive effect of CD4 + CD25 + T cells on autologous T-cell proliferation.
FoxP3 , a forkhead transcription factor family member encoded on the X chromosome, is a critical control gene for the development and function of natural CD4 + CD25 + T-reg cells. In lung cancer cells, tumor-derived cyclooxygenase-2 (COX-2)/PGE 2 induced expression of FoxP3 and increased T-reg cell activity. In vivo, inhibition of COX-2 reduced T-reg activity, attenuated FoxP3 expression in TILs, and decreased tumor burden. In stage I to III NSCLC, tumor-infiltrating FoxP3 + T-regs correlated with COX-2 expression and an increased tumor recurrence.
Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) are a phenotypically heterogeneous population of cells of myeloid origin that expand during cancer, inflammation, and infection, characterized by their immature state and ability to suppress T-cell responses. Unlike the physiologic state where immature myeloid cells generated in bone marrow quickly differentiate into mature granulocytes, macrophages, or DCs, in cancer, a partial block in the differentiation of immature myeloid cells into mature myeloid cells results in an expansion of MDSCs. MDSCs express the common myeloid marker CD33, but lack the expression of markers of mature myeloid and lymphoid cells and the MHC class-II molecule HLA-DR11. Two main subsets of MDSCs that suppress antigen-specific T-cell proliferation to an equal extent, despite their different mechanisms of action, have been identified: a granulocytic subset and a monocytic subset. The MDSC population is influenced by several different factors. The factors that are known to induce MDSC expansion in lung cancer include granulocyte-macrophage colony-stimulating factor, granulocyte colony-stimulating factor, and prostaglandins. The signaling pathways in MDSCs that are triggered by most of these factors converge on Janus kinase protein family members and signal transducer and activator of transcription 3 (STAT3), which are involved in cell survival, proliferation, differentiation, and apoptosis.
The immune-suppressive activity of MDSC is thought to be a result of several mechanisms including upregulated expression of immune-suppressive factors, such as arginase and inducible nitric oxide synthase, and an increase in the production of NO and reactive oxygen species. Arginase, which metabolizes l -arginine to l -ornithine, plays an important role in T-cell suppression through depletion of arginine, which is required for T-cell proliferation and cytokine production. Using the 3LL mouse lung carcinoma model, Rodriguez et al. showed that arginine depletion in the microenvironment by arginase I–producing MDSCs inhibited T-cell receptor CD3 expression and blocked T-cell functions. Arginase I in MDSC was induced by COX-2, and inhibition of COX-2 blocked arginase I induction in vitro and in vivo. Furthermore, blocking arginase I expression using COX-2 inhibitors elicited a lymphocyte-mediated antitumor response.
Further evidence supporting the immunosuppressive effect of MDSCs is derived from experiments of antibody-mediated depletion of MDSC in 3LL tumor-bearing mice. MDSC depletion increased APC activity and augmented the frequency and activity of NK and T-cell effectors that led to reduced tumor growth, enhanced therapeutic vaccination responses, and conferred immunologic memory. MDSC numbers are increased in the peripheral blood of patients with NSCLC compared with healthy volunteers and these cells are capable of directly inhibiting antigen-specific T-cell responses. As with many other cancers, MDSC levels correlate with clinical cancer stage and response to therapy in NSCLC. In patients with advanced NSCLC, higher peripheral blood MDSC numbers were associated with poor response to cisplatin-based chemotherapy and predicted shorter progression-free survival. In patients with early-stage NSCLC, peripheral blood MDSC numbers declined after removal of the tumor. A negative association existed between the population of circulating MDSCs and the frequency of CD8 + T lymphocytes. In a small study of patients with advanced NSCLC who received erlotinib, circulating MDSCs were substantially decreased in patients who had a partial response compared with patients who had progressive disease, and there was a negative correlation between the progression-free survival and MDSC numbers. The prognostic significance of MDSCs in the tumor microenvironment of patient samples in lung cancer is not clear.
MDSC gene expression varies in different tumor types, and an understanding of its clinical significance requires the full characterization of these cells. Despite the lack of consensus over the definition and phenotype of MDSCs, and considerable heterogeneity in how they are defined clinically, their immunosuppressive properties are clear in NSCLC. The mechanisms by which MDSC functions are regulated within the tumor microenvironment, and how they differ from MDSCs that operate at peripheral sites, remain unclear.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


