INTRODUCTION
SUMMARY
The human leukocyte antigens (HLAs) are highly polymorphic glycoproteins encoded by the major histocompatibility complex on chromosome 6. Their biologic function is presentation of antigenic peptides to T lymphocytes, and there are two major classes: class I (A, B, and C loci) and class II (DR, DQ, and DP loci). Class I antigens are present on almost all nucleated cells, whereas class II antigens are primarily expressed on B cells and other antigen-presenting cells such as dendritic cells, endothelial cells, and monocytes. These antigens play key roles in hematopoietic cell transplantation acceptance/rejection and allosensitization to nonleukoreduced blood transfusions leading to platelet transfusion refractoriness, with lesser, but distinct roles in solid-organ transplantation. Other clinically important lineage-specific white cell antigens include those on neutrophils, which are much less polymorphic and less commonly a cause of clinical problems than the HLA system. Antibody to neutrophil antigens plays a role in autoimmune neutropenia, and reactions such as transfusion-related acute lung injury. Platelets also possess a relatively limited number of polymorphic antigens that are involved in clinical problems such as posttransfusion purpura and platelet transfusion refractoriness, and neonatal problems such as alloimmune thrombocytopenia.
Acronyms and Abbreviations
CDC, complement-dependent cytotoxicity; ELISA, enzyme-linked immunosorbent assay; GP, glycoprotein; GVHD, graft-versus-host disease; HLA, human leukocyte antigen; HNA, human neutrophil antigen; HPA, human platelet antigen; MHC, major histocompatibility complex; NAIT, neonatal alloimmune thrombocytopenia; NMDP, National Marrow Donor Program; PCR, polymerase chain reaction; PRA, panel reactive antibody; PTP, posttransfusion purpura; SSO, sequence-specific oligonucleotide; SSP, sequence-specific primer; TRALI, transfusion-related acute lung injury; WHO, World Health Organization.
HUMAN LEUKOCYTE ANTIGENS (MAJOR HISTOCOMPATIBILITY COMPLEX)
The human leukocyte antigens (HLAs) are highly polymorphic glycoproteins encoded by a region of genes known as the major histocompatibility complex (MHC) located on chromosome 6p21 and covering a region of approximately 7.6 Mbp.1,2 After ABO antigens, HLA antigens are the major barrier to transplantation. Their biologic function is to present antigenic peptides to T lymphocytes. The MHC codes for several groups of antigens. The best understood are the highly polymorphic, classical class I (HLA-A, HLA-B, and HLA-C) and class II (HLA-DR, HLA-DQ, and HLA-DP) antigens. Class I antigens are ubiquitous and present on most nucleated somatic cells. Class II antigens exhibit more restricted distribution, with varying levels of expression on B cells, dendritic cells, monocytes, macrophages, and endothelial cells. However, class II antigens can be induced on many cell types through activation.3 The nonclassical class Ib antigens HLA-E, HLA-F, and HLA-G, and the MHC class I chain-related antigens are much less polymorphic, their function less understood, and their tissue expression more limited. In addition the MHC region codes for a number of pseudogenes. This chapter focuses on the classic class I and II molecules because of their importance in transfusion and transplantation.
The two major classes of HLA antigens are homologous. However, there are areas of high variability (polymorphism) that distinguish individual HLA molecules (alleles) and confer antigen specificity. HLA antigens are codominantly expressed so that each individual expresses two antigens at each locus (A, B, DR, etc.). As of July 2014, many thousands of HLA alleles had been characterized.5 Table 137–1 lists the number of known HLA alleles at each locus.
The first sequence map of the MHC encompassed approximately 3.6 Mbp on chromosome 6p21 and was divided into three regions: class I, class II, and class III genes.2 Newer analysis confirming high linkage disequilibrium and conserved synteny led to the concept of an extended MHC (xMHC), and a new map was produced in 2004.6 The xMHC occupies approximately 7.6 Mbp, and is composed of five subregions, which include the classical classes I, II, and III genes. Class II genes are the most centromeric and occupy approximately 1 Mbp of DNA. The genes are ordered sequentially beginning with HLA-DP genes followed by HLA-DM, TAP, HLA-DQ and lastly the HLA-DR genes. The class III genes occupy space between the class I and class II genes. The class III genes include genes that encode other proteins that participate in immune response such as complement, heat shock proteins, tumor necrosis factor, and other lymphocyte antigens. Telomeric are the class I genes sequentially as MICA, MICB, HLA-B, HLA-C, HLA-E, HLA-A, HLA-F, and HLA-G. Extended class I genes include histone clusters and zinc finger genes. Figure 137–1B is a representative map of the MHC.
Figure 137–1.
A. Schematic of the HLA-A2 molecule. The peptide groove is formed by the α helices and β-pleated sheet floor. The groove holds processed peptide antigen. The peptide and the polymorphic α helices interact with the T-cell receptor. B. Representative diagram of the genes of the MHC on chromosome 6. (A, reproduced with permission from Bjorkman PJ, Saper MA, Samraoui B, et al: The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature 329(6139):512–518, 1987. B, adapted with permission from Campbell RD, Trowsdale J: Map of the human MHC, Immunol Today 14(7):349–352, 1993.)
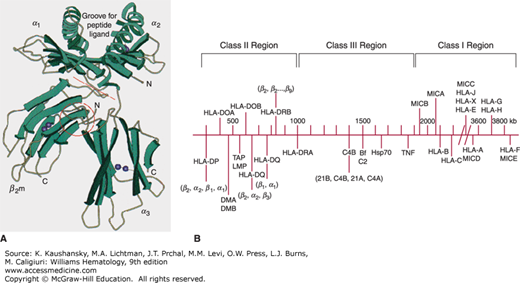
The HLA-A, -B, and -C molecules are transmembrane glycoproteins with an Mr 56,000.7 Each is a heterodimer composed of one α heavy chain (Mr 45,000) noncovalently bound to β2-microglobulin (Mr 11,000). The α heavy chain is the polymorphic glycoprotein encoded by the MHC genes. The extracellular region of the α chain consists of three domains (α1, α2, α3) based on folding and disulfide bonding (see Fig. 137–1A). Antigenicity resides in the α1 and α2 domains, the areas of highest polymorphism. These two chains form a platform composed of a single β-pleated sheet “floor” topped by two α helices with a cleft or groove between them. The structure is supported by the third, α3, domain of the heavy chain in conjunction with β2-microglobulin, which stabilizes the molecule on the cell surface. Class I HLA molecules present peptide fragments from endogenously derived proteins (e.g., viral infection, intracellular bacteria, or transformation) to CD8+ T cells. The highly polymorphic groove permits presentation of highly variable peptides of nine amino acids average length. Class I HLA-A, -B, and -C antigens are found on most nucleated somatic cells.8 Platelets express HLA-A antigens, but lack some HLA-B and most HLA-C antigens.9
The class II antigens are also transmembrane glycoproteins formed by two noncovalently bound chains.12 Both the α heavy chain (Mr 34,000) and the light β chain (Mr 29,000) are encoded in the MHC region. Class IImolecules, like class I, consist of an extracellular hydrophilic NH2– terminal region, a hydrophobic transmembrane region, and an intracellular COOH– terminus region. Unlike class I antigens, the extracellular regions of each chain contain only two domains. The two domains of the α chain are designated α1 and α2, and the two domains of the β-chain are called β1 and β2. The α chain of HLA-DR is constant for all HLA-DR molecules, whereas the β chain is polymorphic and determines specificity of the molecule. Both α and β chains of HLA-DQ and -DP are polymorphic, although the β chain is more so than the α chain. In all class II antigens the β1 domain represents the most polymorphic region. The structure of HLA-DR is essentially similar to the structure of class I molecules. Class II antigens present peptides from exogenous sources, such as bacterial pathogens, to CD4+ cells. The binding groove is more open than that of class I, and peptides of longer length (11 to 18 amino acids) are accommodated.13,14 Class II antigens have a more restricted tissue distribution, being found primarily on B lymphocytes and other antigen-presenting cells such as dendritic cells, monocytes, and macrophages. They may also be expressed on activated endothelial cells and T lymphocytes.12
The extraordinarily polymorphic nature of HLA has probably evolved because of the need to present a very large array of different antigenic peptides in host defense. Antigen processing and presentation is a tightly regulated process, especially among the professional antigen-presenting cells such as dendritic cells. A number of alternative mechanisms have been demonstrated in vitro, such as cross-presentation, whereby dendritic cells transfer antigen derived from endocytic sources to the class I pathway, but are poorly understood.15 One promising area of research is the ability of HLA molecules to present antigenic peptides derived from tumors. Such peptides could arise via point mutation, or reactivation of a normally silent gene that produces a peptide that can bind to HLA and induce a T-cell immune response. Several such peptides (melanoma-associated gene [MAGE] antigens) have been identified for melanoma.16
Distinguishing polymorphic variations among HLA antigens is clinically important in stem cell transplantation. Terminology used to describe accepted HLA alleles or antigens is standardized by the World Health Organization (WHO), Nomenclature Committee for Factors of the HLA System, which issues biannual reports and monthly updates.4 In addition, an HLA dictionary defining HLA antigens, their assigned nomenclature, and serologic equivalents is published periodically.5 The nomenclature committee approved major changes to the system that were implemented in 2010.17 The revisions were designed to accommodate the unexpected number of new sequenced alleles. Under this system, colons are used as delimiters to separate fields. The first field signifies the allele family that often corresponds to the serological antigen. The second field denotes the alleles, assigned in order of determination. The third field is used for defining synonymous nucleotide substitutions. The last field defines alleles that differ by sequence polymorphisms in introns or in the 5′ or 3′ untranslated regions that flank the exons and introns. In addition, there are suffixes that are used to describe expression status. Null alleles (not expressed) are identified by the suffix “N.” Low surface expression is represented by “L.” Secreted molecules not present at the surface are assigned “S.”
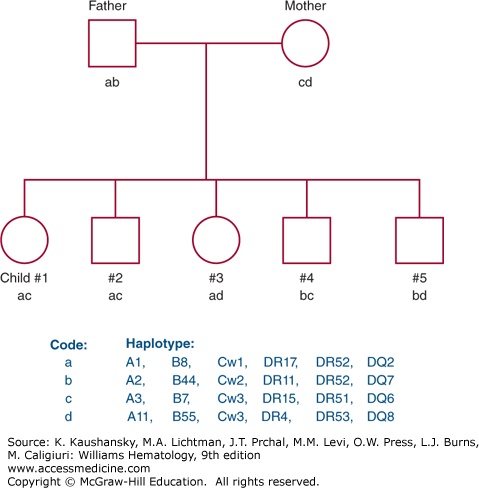
The genes of the MHC demonstrate more polymorphism than any other genetic system; that is, many alleles exist for each locus. Each individual, however, has one allele for each locus per chromosome, and therefore, encodes two HLA antigens per locus. The identification of each HLA antigen of an individual is called a phenotype. Because HLA genes are closely linked, recombination within the MHC is rare (≤1%), and a complete set of HLA genes usually is inherited from each parent as a unit. The genes inherited from each parent are referred to as a haplotype. Maternal and paternal haplotypes can be identified through family studies. Identification of both haplotypes of an individual provides the genotype. Family studies consist of typing for the HLA-A, HLA-B, HLA-C, HLA-DR, and HLA-DQ antigens to identify haplotypes and to rule out genetic recombination within the MHC. Because HLA genes are inherited together on a single chromosome, four combinations of maternal and paternal haplotypes are possible provided no recombination occurs (Fig. 137–2).
Figure 137–2.
Pedigree representing inheritance of HLA antigens. Each of the four parental haplotypes is coded by a letter: a and b represent paternal haplotypes; c and d represent maternal haplotypes. Each child inherits one paternal and one maternal haplotype such that four combinations are possible.
Because the MHC is so highly polymorphic, the probability that any two unrelated individuals are HLA identical is extremely low. However, the system exhibits a phenomenon known as linkage disequilibrium. That is, HLA alleles are inherited together on the same chromosome more often than would be predicted if HLA loci were in equilibrium. At equilibrium, the frequency of an allele at one locus is independent of the frequencies of alleles at linked loci. For example, the gene frequency of HLA-A1 is 0.145 and that of HLA-B8 is 0.1 in North American whites. Given no preferential association between A1 and B8, then the haplotype frequency would be 0.0145 (0.145 × 0.1). However, population studies demonstrate that the actual frequency of the HLA-A1, B8 haplotype is 0.0726.18 The degree of linkage disequilibrium is defined as the observed frequency minus its expected frequency, 0.0581 in this example. Although particular alleles found in linkage disequilibria differ for various racial groups, all racial groups display significant disequilibria. Different races and ethnic groups can vary greatly in the frequency with which HLA antigens are found.19
Tissue typing for HLA antigens can be performed by various methods using serologic, cellular, and molecular technologies. The most frequent procedures used in the clinical setting are serologic and molecular. Cellular assays such as the mixed lymphocyte reaction and the primed lymphocyte test were common prior to the widespread adoption of DNA methods. Compared to DNA techniques, cellular methods are labor-intensive and require the use of radioisotopes; they are mainly used in research laboratories.
The microlymphocytotoxicity complement-dependent cytotoxicity (CDC) test has been a fundamental procedure for defining HLA antigens for more than 30 years,20 although it has been supplanted by molecular typing methods. In this assay, a suspension of lymphocytes is incubated with human alloantisera or monoclonal antibody in a microtiter tray.21 Rabbit serum is added as a source of complement. Cell death is induced when antibody binds to antigen on the cell surface and the complement cascade activated. Death is visualized microscopically by the uptake of vital dye or by immunofluorescence. Panels used to determine a patient’s HLA type consist of two to four antisera that recognize the same specificity, which requires approximately 150 different reagents for class I antigens and 80 to 150 for class II antigens. Antisera are usually obtained from multiparous women, multiply transfused patients, and from patients who have rejected allografts. Monoclonal antibodies are also commercially available for many HLA specificities. Serology for class II (DR and DQ) antigens requires enrichment for B lymphocytes, which can be accomplished with antibody or immunomagnetic bead reagents.
The development of the polymerase chain reaction (PCR)22 revolutionized the approach to HLA typing. Several DNA-based methods are commonly accepted for HLA typing. These include sequence-based typing, sequence-specific primer (SSP) amplification23 and sequence-specific oligonucleotide (SSO) probe hybridization. All of these methods involve amplification of genomic DNA from selected portions of HLA genes with oligonucleotide primer pairs. Generally exons 2 and 3 of class I and exon 2 of class II genes are amplified. These exons encode most of the polymorphisms of the classes I and II molecules. Molecular HLA typing is primarily of clinical interest in marrow/blood stem cell transplantation.
The advent of “next-generation sequencing” (NGS) strategies has proven useful to high-throughput sequencing of HLA genes. NGS methods also overcome limitations of Sanger-based methods, including combination ambiguities that result from heterozygous samples in diploid genomes or between alleles where sequence varies outside the target region (i.e., exons 2 and 3). NGS methods are described as massively parallel allowing for many overlapping reads of the same sequence area. Multiple platforms using different chemistries are available; bioinformatics expertise is required to analyze the extensive data provided by these methods.
In addition to typing for HLA antigens, most laboratories also use technology to detect antibodies to HLA antigens. This is very important for solid-organ transplantation where the presence of anti-HLA antibodies can cause irreversible rejection upon transplantation. It is of less concern for marrow/stem cell transplantation where donors and recipients are generally matched for HLA antigens. The microcytotoxicity serologic test is still in use, but solid phase assays have become standard practice as they are more sensitive than the CDC method. These assays include enzyme-linked immunosorbent assay (ELISA) and microbead-based flow assays such as FlowPRA and Luminex assays. These tests require HLA antigen, in either recombinant or native form, bound to a solid surface such as a microsphere and used to capture alloantibody in patient serum. Analysis of the reaction patterns yields information about the breadth of alloimmunization, or PRA (panel reactive antibody), and the specificity of the reactions. Prior to most solid-organ transplants, a donor-specific crossmatch is also performed to ensure that the recipient does not have anti-HLA antibodies against donor HLA antigens. Crossmatches are performed by microlymphocytotoxicity (CDC), flow cytometry, and by solid-phase (ELISA and microbead) assays. Labs are now adopting the “virtual” crossmatch using data from the sensitive microbead assays to predict crossmatch outcome. For low-risk cases this allows a transplant to proceed without waiting for a physical crossmatch and shortens cold ischemic time for an organ.24
The HLA antigens coded by the MHC play a central role in transplantation, regulation of immune responses, and susceptibility to a variety of diseases. The most common application, however, is the field of transplantation. In renal and stem cell transplantation, allografts from HLA-identical sibling donors have significantly greater survival than grafts from nonmatched family or unrelated donors.
For solid-organ transplantation, a living donor is not always available or feasible (e.g., for heart transplantation). HLA typing for matching of kidneys and pancreas is performed at the HLA-A, HLA-B, and HLA-DR loci at low resolution (serologic or antigen level by DNA). In the early years of renal transplantation a high degree of match was sought between recipients and donors. However, as more potent immunotherapies have developed, the level and use of HLA matching has declined. HLA matching is not prospectively performed for hepatic or cardiac transplantation. Detection of alloantibody by screening techniques and the donor specific crossmatch is of prime importance for kidney and heart transplants where its existence could cause a hyperacute rejection and graft failure. The role of alloantibody is less clear in the immediate posttransplantation period, but may be detrimental to long-term survival.25 In the last several years, several “paired donor exchanges” have arisen to assist recipients who have incompatible, but willing living donors. These programs, such as the National Kidney Registry allow donor-recipient pairs to swap within or between transplantation centers. Chains of up to 30 cross-country swaps in the United States have facilitated more than 1200 transplantations since its inception.
Marrow or stem cell transplantation entails problems other than allograft survival. In these therapies an immunocompetent graft is transplanted to an immunocompromised/immunoablated host. The graft may recognize the host tissue as foreign and mount an immune response resulting graft-versus-host disease (GVHD). With HLA-identical sibling donors, disease-free survival of greater than 80 percent can be achieved for some hematopoietic malignancies.26,27 However, fewer than 30 percent of individuals have an HLA-identical sibling. For these patients, alternative donors, such as phenotypically matched unrelated volunteers and partially matched family members, may be considered. However, the risks and incidence of graft failure and GVHD are higher than seen with HLA-identical siblings, and increase with the level of HLA disparity. HLA typing for stem cell transplants is generally performed by molecular methods. For those with a family donor, low-resolution typing may be sufficient to identify a match. However for unrelated or haploidentical family donors, high-resolution (allele-level) typing for HLA-A, HLA-B, HLA-C, HLA-DR, and HLA-DQ should be performed, and is required by the national registry program (National Marrow Donor Program [NMDP]).26 HLA alloantibody is becoming common, especially when incompletely matched donors are used.
Patients requiring platelet transfusions may be broadly sensitized to HLA-A, and -B (i.e., have high PRA) through prior transfusions (particularly nonleukoreduced) or pregnancies. HLA antibody screening to select nonreactive donors and/or HLA donor platelet matching may enable these refractory patients to achieve improved platelet transfusion count increments.
HLA typing at one or a few antigens or alleles may also be performed to support diagnosis of diseases associated with specific HLA antigens. The most common of these is the association between HLA-B27 and ankylosing spondylitis28 and HLA-DQ2’s association with narcolepsy.29 HLA typing may also be performed to determine eligibility for vaccine trials that use peptides and HLA.30,31 HLA antigens also are implicated in drug hypersensitivity. For example, HLA-B*5701 is associated with hypersensitivity to the drug for treatment of the human immunodeficiency virus, abacavir.32
HLA tetramers may be used to monitor the efficacy of HLA-based peptide vaccines. Recombinant HLA molecules are loaded with the peptide vaccine and linked via a fluoresceinated streptavidin molecule. They are incubated with patient blood lymphocytes. Effector T cells specific for the peptide-HLA will be bound by the tetramer and monitored by flow cytometry.
NEUTROPHIL ANTIGENS AND ANTIBODIES
Clinically significant alloantigens expressed only or predominantly by neutrophils are known as human neutrophil antigens (HNAs).33 In this nomenclature, the antigen systems are indicated by integers, and specific antigens within each system are designated alphabetically by date of publication (Table 137–2).
| System | Alleles | Location of Antigens | Genes |
|---|---|---|---|
| HNA-1 | HNA-1a, -1b, -1c, and 1d | FcγRIIIb | FCGR3B*01, FCGR3B*02, and FCGR3B*03 |
| HNA-2 | HNA-2 | NB1gp | CD177 |
| HNA-3 | HNA-3a | Choline transporter-like protein-2 (CTL2) | SLC44A2*01 |
| HNA-4 | HNA-4a | αM integrin, C3bi-receptor (CR3)(CD11b) | ITGAM*01 |
| HNA-5 | HNA-5a | αL integrin, LFA-1(CD11a) | ITGAL*01 |
The neutrophil-specific HNA-1 antigen system is made up of the four antigens alleles, HNA-1a, -1b, -1c and -1d (see Table 137–2).34 HNA-1 antigens are located on the low-affinity Fcγ receptor IIIb (FcγRIIIb), CD16, and are expressed only on neutrophils.35,36,37,38 FcγRIIIb and HNA-1 antigens are expressed on all segmented neutrophils, on approximately one-half of neutrophilic metamyelocytes, and on approximately 10 percent of neutrophilic myelocytes.39 Soluble FcγRIIIb is present in plasma and has the same HNA-1 polymorphisms found on neutrophils.40
FcγRIIIb and the HNA-1 antigens are encoded by the FCGR3B gene located on chromosome 1q23–24. FCGR3B is highly homologous to FCGR3A, which encodes FcγRIIIa. In addition to the polymorphic FCGRB3B nucleotides only four others differ between FCGR3B and FCGR3A. The most important difference between the two genes is a C-to-T change at 733 in FCGR3B that creates a stop codon. As a result, FCGR3A has 21 more amino acids than FCGR3B and FCGR3B is a glycosylphosphatidylinositol (GPI)-anchored rather than a transmembrane glycoprotein (GP).
The four HNA-1 system antigens are encoded by three alleles. The FCGR3B*01 allele differs from FCGR3B*02 by only five nucleotides in the coding region, at positions 141, 147, 227, 277, and 349.35,36,37,38 Four of the nucleotide changes result in changes in amino acid sequence between the HNA-1a and HNA-1b forms of FcγRIIIb. The glycosylation pattern of FcγRIIIb differs between the two antigens because of two nucleotide changes at bases 227 and 277. The HNA-1b form of FcγRIIIb has six N-linked glycosylation sites and the HNA-1a form has four glycosylation sites.
The FCGR3B*03 allele is identical to FCGR3B*02 except for a C-to-A substitution at nucleotide 266 resulting in an alanine to aspartate change at amino acid 78 of FcγRIIIb.34 In many cases, FCGR3B*03 exists on the same chromosome with a second or duplicate FCGR3B gene.41,42 FCGR3B*03 encodes both HNA-1b and HNA-1c. FCGR3B*02 also encodes HNA-1d which is characterized by the FcγRIIIb sequence Ala78–Asn82.43
The antigen frequencies of three of the alleles vary widely among different racial groups. Among whites, HNA-1b is the most common antigen (Table 137–3),44,45,46,48,49,50 but in Japanese and Chinese populations HNA-1a is most common.44,46,48,49,51 The frequency of the gene encoding HNA-1c antigen also varies among racial groups. HNA-1c is expressed by neutrophils in 4 to 5 percent of whites, 25 to 38 percent of African Americans, and 10 percent of Brazilians.52,53 The antigen frequency of HNA-1d should be nearly the same as that of HNA-1b, but it has not yet been investigated.
| Antigen Frequencies | |||||
|---|---|---|---|---|---|
| System | Alleles | Europeans/North Americans | Africans and African Americans | Asians | Brazilians/Argentineans |
| HNA-1 | HNA-1a | 58% | 59–67% | 91% | 68% |
| HNA-1b | 88% | 71–88% | 54% | 76% | |
| HNA-1c | 5% | 25–38% | <1% | 5–10% | |
| HNA-1d | NA | NA | NA | NA | |
| HNA-2 | HNA-2 | 95–97% | 95% | 89–99% | NA |
| HNA-3 | HNA-3a | 89–92% | NA | NA | NA |
| HNA-4 | HNA-4a | 99% | NA | 99% | 97% |
| HNA-5 | HNA-5a | 85% | 88% | 81–96% | 78–92% |
Several other sequence variations in FCGR3B have been described.46 Most of these chimeric alleles have single-base substitutions involving one of the five single nucleotide polymorphisms (SNPs) that distinguish FCGR3B*1 and FCGR3B*2. FCGR3B alleles that most closely resembled FCGR3B*2 were found more often in African Americans than in whites or Japanese.46,47
Genetic deficiencies of neutrophil FcγRIIIb and HNA-1 antigens have also been reported. Among whites the incidence of individuals homozygous for FCGR3B deletion is approximately 0.1 percent.52,54,55 However, among Africans and African Americans the incidence is much higher. In one study, 3 of 126 Africans were found to be FCGR3B deficient,48 and in another study, 1 of 53 were found to be FCGR3B deficient.46
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


