Abstract
This chapter is an overview on breast cancer, prostate cancer, and endometrial cancers, with an emphasis on hormone-related components. We review the epidemiology of these cancers, focusing on the hormonal risk factors. The majority of the chapter is dedicated to a review of the main biological roles of the hormone receptors, estrogen receptor, and progesterone receptor for breast cancer, and endometrial cancer and androgen receptor for prostate cancer, in tumor initiation and as targets of endocrine treatments. We also discuss in detail the types of endocrine treatments, mechanisms of resistance to endocrine treatments, and approaches to overcome resistance.
Keywords
Breast cancer, estrogen receptor (ER), progesterone receptor (PR), endocrine treatments, resistance to endocrine treatments, genomic aberrations, prostate cancer, androgen receptor (AR), prostate-specific antigen (PSA), endometrial cancer
Breast Cancer
- ◆
Breast cancer is the most common cancer in women.
- ◆
Breast cancers are molecularly classified to luminal A, luminal B, basal-like, and HER2 enriched breast cancers. Clinically breast cancers are classified to hormone receptor positive, HER2 positive, or triple negative. The majority of breast cancers are hormone receptor positive.
- ◆
Despite effective endocrine treatments that target the estrogen receptor (ER), resistance to these treatments is a major clinical challenge. There are multiple mechanisms for endocrine resistance. CDK4/6 inhibitors have recently been approved for the treatment of hormone receptor positive breast cancer.
Worldwide breast cancer is the most common cancer diagnosed in women and the leading cause of cancer mortality in women. In the United States, breast cancer is the most common cancer diagnosed in women and the second leading cause of cancer mortality after lung cancer. It is estimated that in 2017 there will be about 252,710 new cases of invasive breast cancer and more than 40,000 women will die from breast cancer in the United States. Much progress has been made in the care of breast cancer over the past 30 years; since 1989, death rates from breast cancer have dropped, and this is likely due to screening programs for earlier detection and improvements in treatments (see American Cancer Society: Cancer Facts and Figs. 2016 ).
Risk Factors
Dietary, Environmental, and Lifestyle Factors
Epidemiological observations suggest a role for high fat diet, alcohol, exercise, and obesity in the development of breast cancer. In Japan, the rate of breast cancer peaks at the age of menopause, but in the United States, incidence continues to increase until age 90. The difference in postmenopausal patterns may result from the increase in obesity and associated aromatase increments in women in the United States compared with Japan. This differential breast cancer rate does not appear genetic since Japanese women who move to the United States experience an increased rate of breast cancer that later approaches that of North American women.
Hormonal Factors
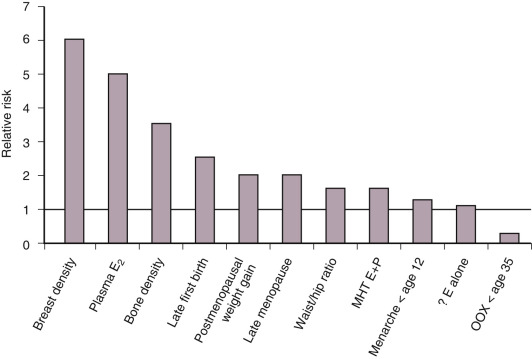
Hormonal factors and particularly estrogens contribute to the development of breast cancer (see Chapter 14 ). Administration of exogenous estrogens to various animal species results in breast neoplasms. Spontaneous development of breast cancer in aging rats can be prevented by oophorectomy or administration of aromatase inhibitors to block estrogen production. As outlined in Fig. 29.1 , epidemiological studies implicate several hormonal factors that are associated with an increased incidence of breast cancer.
Intensity of Estrogen Exposure
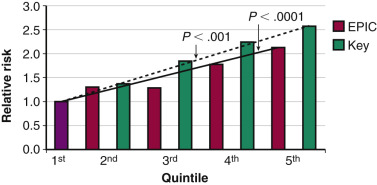
The majority of risk factors for breast cancer relate to the duration or intensity of a woman’s exposure to endogenous or exogenous estrogens (see Fig. 29.1 ). Early menarche and/or late menopause increase breast cancer risk. Elevations in circulating estradiol levels predict the risk of developing breast cancer over the ensuing years in postmenopausal women ( Fig. 29.2 ). Estrogen levels provide information independent of other known risk factors. An estradiol in the top quintile may increase the relative risk (RR) of breast cancer by threefold to fivefold (see Fig. 29.2 ). An index weighing several circulating estrogens (E1, E2, E3, E1-S, E2-S) provides a higher estimate of risk than estradiol alone. Estrogen metabolites, which do not act through the ER, are also associated with an increased risk of breast cancer. For example, weighted averages of several estradiol metabolites involving the 4-hydroxylation pathway predict a greater risk of breast cancer than levels of E2 alone.
Putative markers of long-term estrogen exposure such as bone density are also predictive. Women in the top quartile of bone density have a threefold increased risk of breast cancer (see Fig. 29.1 ); a history of fracture or height loss lowers the risk. Gain of at least 20 kg as an adult increases breast cancer risk twofold, and weight loss reduces the risk. Increased waist-hip ratio exerts a similar increase in risk. Combined analyses of multiple studies indicate that alcohol intake can increase the risk of breast cancer, perhaps by decreasing the clearance of estradiol. Increased exposure to estradiol in utero, as shown by twin studies, may increase the risk of breast cancer by as much as twofold. Early pregnancy and prolonged duration of breastfeeding diminish the risk. More dramatic is the 75% reduction in risk caused by bilateral oophorectomy before age 35 or the use of antiestrogens by premenopausal and postmenopausal women.
Sources of Estrogen
The estradiol present in breast tissue is synthesized in three sites: the ovary, extraglandular tissues, and the breast itself. Direct glandular secretion by the ovary results in delivery of estradiol to the breast through an endocrine mechanism in premenopausal women. After menopause, the extraglandular production of estrogen from ovarian and adrenal androgens in fat and muscle provides the second source of estradiol. Thirdly the breast itself can synthesize estradiol via aromatization of androgens to estrogens, cleavage of estrone-sulfate to free estrone via the enzyme sulfatase, and conversion of estrone to estradiol via 17βOH-HSD. Classic studies involving infusion of radiolabeled androgens and estrogens into women provide direct evidence of both in situ production in breast and uptake from plasma. Recent studies relating plasma estrogen levels to expression of estrogen responsive genes in breast tumor tissue suggest that ER mediated uptake might predominate over in situ synthesis. Several factors regulate in situ estradiol synthesis, but the most important is the degree of obesity, which increases the amount of aromatase in breast and, consequently, estradiol production. The mechanism for obesity-related induction of aromatase involves leptin, adiponectin, and AMP-kinase, which allow CRTC2 to become detached from a 14-3-3 protein and enter the nucleus to increase aromatase transcription.
Mammographic Density
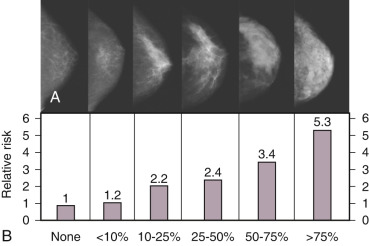
Breast density represents a strong risk factor for breast cancer risk ( Fig. 29.3 ). Increase in breast cancer risk from lowest to highest breast density category is on the order of fivefold, depending upon the age of the patient (see Fig. 29.3 ), with greater RR in older women. Mammographic density, when added to the factors used in the Gail predictive model, increases the power of prediction and thus is independent of several other risk factors. Radiographic mammographic density corresponds to a higher fraction of stroma and epithelium relative to adipose tissue and that areas of increased breast density contain higher levels of aromatase than nondense areas, suggesting a role for increased in situ estrogen production. The cause(s) of increased breast density are poorly understood but are partly related to genetic factors as shown by twin studies and estrogen levels, since exogenous estrogens increase and antiestrogens reduce breast density. Only those individuals with a reduction in breast density of greater than 10% on tamoxifen experienced prevention of breast cancer, further supporting the biological significance of breast density.
Exogenous Hormones and Breast Cancer Risk
In premenopausal women, the use of oral contraceptives for 10 or more years increases the RR of breast cancer by approximately 10% to 20%. This increase affects very few women, since the age-related absolute incidence of breast cancer is minimal in women taking oral contraceptives. The median age in the largest study of oral contraceptive use was 26 years old. Based on Surveillance Epidemiology and End Results (SEER) population incidence data for 25- to 30-year-old women (approximate incidence 12/100,000 per year), use of an oral contraceptive would result in only 1.2 to 2.4 extra breast cancers per 100,000 women per year.
The concept that menopausal hormonal replacement treatment (MHT) increased the risk of breast cancer was initially controversial, as it was based on conflicting observational studies. However, the large Collaborative Group on Hormonal Factors in Breast Cancer Study (CGHFBC study), which was a meta-analysis of 51 studies published in 1997, helped clarify some of the factors responsible for the differing conclusions among the various observational reports, as outlined here :
- 1.
The RR of breast cancer from MHT is small, and large studies with a long duration of follow-up are required to minimize type I and type II statistical errors.
- 2.
The risk of breast cancer increases linearly with duration of MHT use. Accordingly, comparisons of “ever users” with “never users” are invalid, since duration of estrogen use is not considered .
- 3.
The increased risk of breast cancer imparted by MHT dissipates within 4 years of cessation of therapy. Therefore only women using MHT within 4 years of study might be found to be at increased risk.
- 4.
Breast cancer risk also diminishes over a 4-year period following menopause, presumably as a reflection of decreased estrogen and progesterone levels. As a result, analyses of observational studies need to match users versus nonusers as to time following menopause.
- 5.
The increased risk of breast cancer from MHT is higher in women with lower weight or body mass index (BMI). With inclusion of a large proportion of women with high BMIs, a single study might then obscure associations between MHT use and breast cancer risk.
When the CGHFBC took these confounding factors into account, the pooled observational data were relatively consistent and suggested an increased risk of breast cancer in women using estrogen plus a progestogen (P) over 5 years or taking estrogen alone long-term for 10 or more years.
The large, prospective, randomized, Women’s Health Initiative (WHI) trial in postmenopausal women first published in 2002 strongly supported the observational data on estrogen plus progestogen (E+P). Nearly 16,000 postmenopausal women with an average age of 63 enrolled in the estrogen plus progestin arm of the WHI study and received either placebo or conjugated equine estrogens (0.625 mg) plus medroxyprogesterone acetate (MPA; 2.5 mg) for 5 years. At study termination after 5.2 years, the RR was reported as 1.26 (RR, 1.26; 95% CI, 1.00 to 1.59). The absolute increase in risk (i.e., attributable risk) was small, with 4 cases per 1000 women treated for 5 years.
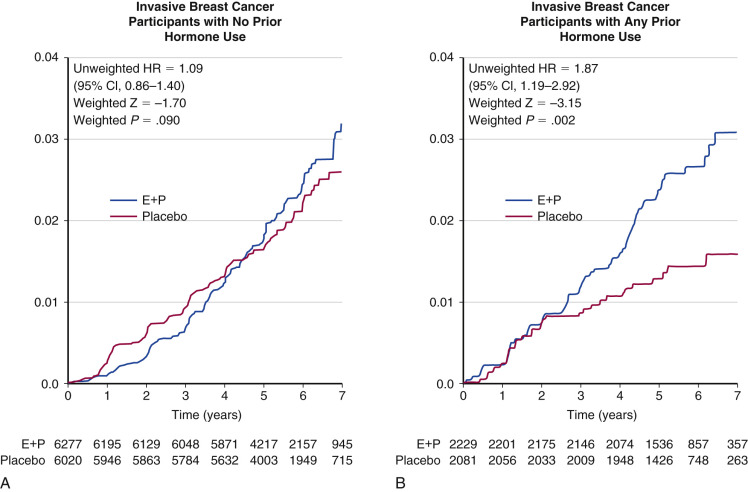
In the original WHI report, it was noted that 74% of the participants were never users of MHT, 19.7% were past users, and 6.4% were current users. Among the never users, breast cancer risk was not increased (RR, 1.09; 95% CI, 0.86 to 1.40), a statistic confirmed in a subsequent publication in 2006 ( Fig. 29.4 ). In another analysis, the risk of MHT was shown to be higher for women who initiated treatment within 5 years of menopause (deemed a short gap time), as opposed to those who initiated after a long interval (long gap time).
Effect of Specific Progestogens and Progesterone
The WHI E+P study utilized only one progestogen (synthetic progesterone)―MPA―and did not address whether different progestogens might have lesser effects on breast cancer risk. Several observational studies examined the effects of various types of progestogen, as well as differences between combined continuous and sequential regimens. The Million Women Study suggested that all types of progestogen (MPA, norgestrel, and norethisterone) are associated with an increased risk of breast cancer, and the combination of an estrogen and progestogen was associated with an approximately fourfold greater increase in breast cancer incidence than estrogen alone. However, there is evidence that progesterone itself may be associated with a lower risk or no risk of breast cancer than the synthetic progestogens. Studies examining the levels of circulating progesterone and risk of breast cancer showed either no association or a decreased risk. In addition, the French component of the European Prospective Investigation into Cancer and Nutrition (EPIC) study reported on 2354 cases of invasive breast cancer among 59,216 French postmenopausal women followed for an average of 8.1 years. This study reported a RR of 1.08 (CI, 0.89 to 1.31) for estradiol in combination with micronized progesterone. In contrast, the RR for estradiol combined with synthetic progestogens in that study was 1.69 (CI, 1.50 to 1.91), similar to risks reported in other epidemiological studies.
An understanding of the physiological basis for the association of progestogens with breast cancer rests on data indicating that progestogens are mitogenic on breast tissue in contrast to their antimitogenic effects on the uterus. While data from cell cultures or animal studies are conflicting, the weight of evidence from patients suggests that progestogens are mitogenic in the breast. Mammographic studies demonstrate that estrogen/progestogen combinations increase breast density to a greater extent than estrogen alone or placebo. Histological examination demonstrates enhanced cell proliferation and percent of the breast containing glandular tissue as a function of duration of progestogen usage. Increased proliferation would be expected to increase both initiation and promotion of breast cancer in a manner similar to that thought to occur with estrogens.
Confounding Factors
Several commentators have pointed out factors confounding the interpretation of the WHI studies: (1) the frequency of dropouts (30%) and drop-ins (7%; i.e., patients randomized to placebo who then decided to take MHT during the study); (2) the exclusive use of MPA as the progestogen; (3) the average age of participants, 63 years; (4) the fact that 26.5% of women had used prior hormone therapy and then underwent washout before starting MHT; (5) the paucity of women with menopausal symptoms; (6) the fact that only 3.5% of the subjects were in the 50- to 54-year-old age group, which represents women who usually consider initiation of MHT use; and (7) limitation of an increase in breast cancer to women who had previously used MHT and then stopped before randomization (see Fig. 29.4 ). Despite these issues, the WHI remains the only large randomized trial, and the data on E+P and breast cancer risk appear valid. Further, the conclusions are congruent with the findings of the majority of the observational studies, as reported in a meta-analysis.
Women’s Health Initiative Estrogen Alone Study
Another arm of the WHI compared placebo with conjugated equine estrogen alone (E alone) in women who had previously undergone a total abdominal hysterectomy. In the 11.8 year follow-up of the WHI E alone trial (i.e., 7 years of E alone and 4.8 years of subsequent observation), the reduction in breast cancer risk did reach statistical significance in the total group (RR, 0.77; CI, 0.62 to 0.95) and to an even greater extent in compliant patients (sensitivity analysis RR, 0.69; CI, 0.49 to 0.95; Fig. 29.5 ). The protective effect was confined to those without a family history of breast cancer or benign breast disease.
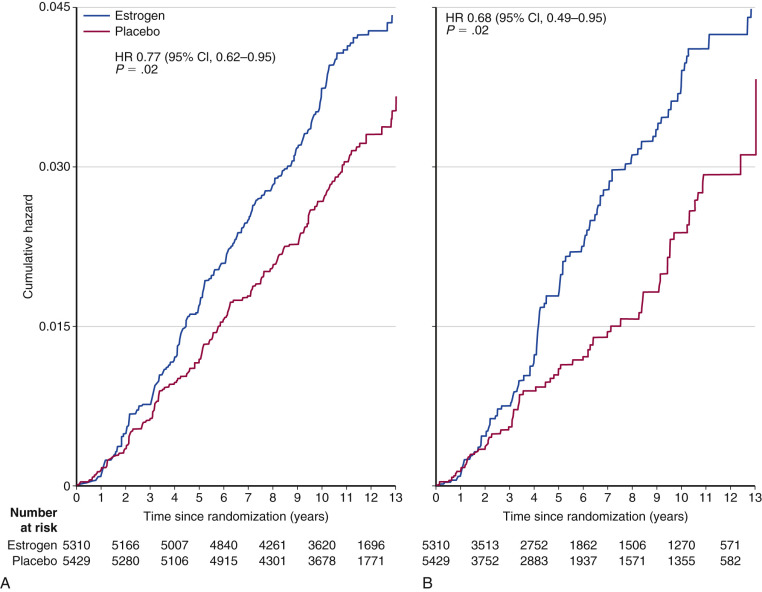
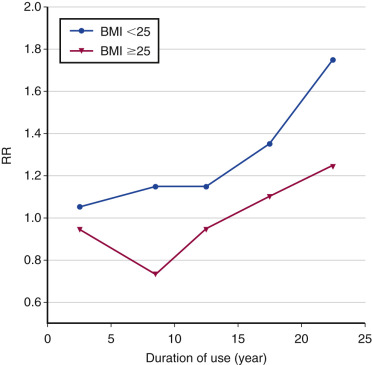
The observational trials reporting an increased risk of breast cancer with long-term estrogen use and the WHI study reporting a reduced risk would seem paradoxical. A plausible explanation for this discrepancy is the “gap time” (i.e., the duration between onset of menopause and start of MHT). Women starting many years after onset of menopause (i.e., “long gap”) experienced a significant reduction in breast cancer risk (RR, 0.58; CI, 0.36 to 0.93), whereas those starting immediately after menopause (“short gap”) did not (RR, 1.12; CI, 0.39 to 3.21 reported initially; RR, 0.89; CI, 0.66 to 1.20 in a later analysis). Data from the Nurses’ Health Study suggest that the increased risk only occurs in women using E alone for more than 10 years, particularly in women with a BMI of less than 25 ( Fig. 29.6 ).
Benign Breast Disease and Risk of Breast Cancer
Benign breast lesions with an enhanced rate of proliferation predict an increased incidence of breast cancer over time ( Fig. 29.7 ). Hyperplastic ductal lesions are often multicentric, suggesting that some type of underlying abnormality is present that predisposes to such lesions. This has been called a field defect or a mutator phenotype . The multifocal nature of the associated benign hyperplastic lesions is most apparent in breast tissue from women with cancer. Examination of tissue adjacent to an invasive breast cancer or in the contralateral breast reveals one or more additional hyperplastic lesions in approximately 40% of patients.
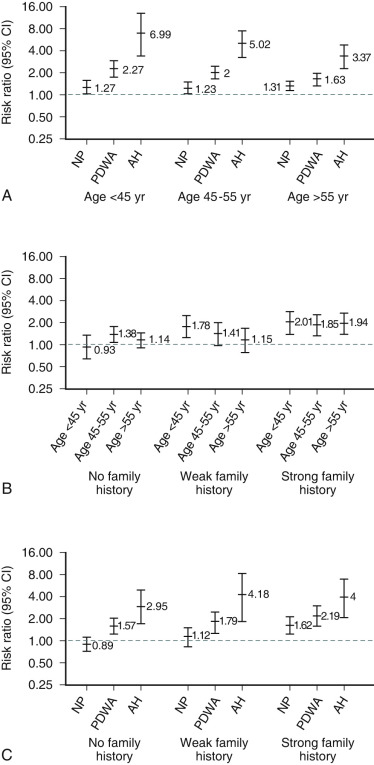
A major consideration for women who present with breast problems is whether they have a higher than normal risk of developing breast cancer. Certain breast lesions such as fibrocystic changes are associated with no increased risk of subsequent breast cancer unless a strong family history is present. Other lesions, characterized by the presence of increased cellular proliferation such as usual ductal hyperplasia, hyperplastic elongated lobular units, and solitary or multiple papillomas impart a small (i.e., less than twofold) relative increase in risk (see Fig. 29.7 ). With atypical ductal hyperplasia (ADH), the RR is 3.88 overall and 10.35 in those with multifocal (i.e., 3 or more) lesions with calcifications. In younger women, ADH imparts a 6.75 RR (i.e., women younger than 45 years old). The RR of development of invasive cancer is increased 10-fold to 12-fold when ductal carcinoma in situ (DCIS) and lobular carcinoma in situ (LCIS) are present.
Proliferative lesions can progress to invasive cancer as evidenced by the increase in ipsilateral breast cancer in women harboring these lesions. However, risk of contralateral breast cancer is also increased when the period of observation extends over more than 10 years, suggesting an underlying defect affecting both breasts. The Mayo Clinic study of nearly 15,000 women established that only lesions with increased proliferation impart an increased risk of breast cancer, unless a strong family history is present.
Estimating Breast Cancer Risk
The Gail model (and more recently the modified Gail model) was developed to aid in assessing breast cancer risk. This model uses answers to seven questions to calculate the 5-year and lifetime risk of developing breast cancer and is used for women 35 years of age or older that do not harbor a BRCA1/2, P53, or PTEN mutation, do not have a strong family history of breast cancer, do not have a history of thoracic radiation before the age of 30, and do not have a history of LCIS. This model has recognized deficiencies in that it does not consider breast density, plasma estradiol levels, bone density, BMI, weight gain in adulthood, second degree relatives with breast cancer, proliferative lesions of breast other than ADH, alcohol intake, or birth control pill and MHT use. Nonetheless, several prospective studies validated the Gail model in high-risk (National Surgical Adjuvant Breast Project [NSABP] prevention study) and in average-risk women (the Nurses’ Health Study). The ratio of observed to expected cancers using this tool was RR 1.03 (95% CI, 0.88 to 1.21) in high-risk women and RR 0.94 (95% CI, 0.89 to 0.99) in average-risk women, and both were highly statistically significant. This risk tool is available on the National Cancer Institute website.
Biological Subtypes of Breast Cancer
For many years the WHO classified breast cancers to 17 histological subtypes. However, this classification was limited, since the majority of breast cancers were classified as invasive ductal carcinoma (IDC) not otherwise specified, and tumors within this class had distinct clinical behavior and response to treatments. The use of the microarray technology led to major advances in the understanding of the molecular heterogeneity of breast cancers, and in seminal studies breast cancers were classified to four intrinsic subtypes. These subtypes include luminal A, luminal B, HER2 enriched, and basal-like breast cancer. Luminal A subtype breast cancers are usually positive for ER and progesterone receptor (PR), have a low pathological grade, and have low proliferation. Luminal B subtype cancers are ER-positive, usually PR negative, have a higher grade and higher proliferation index, and are less responsive to endocrine treatment. HER2-enriched tumors have HER2 (ERBB2 ) amplifications and high pathological grade. Basal-like cancers typically do not express hormone receptors or HER2, are characterized by the expression of basal markers, such as CK5 and CK6, and have a high pathological grade and proliferation index. The intrinsic subgroup of basal-like cancers is a heterogeneous class of cancers by itself and can be further classified to six subtypes, including two basal-like subtypes, an immune-modulatory subtype, a mesenchymal subtype, a mesenchymal stem-like subtype, and a luminal androgen-receptor subtype. Subsequent studies showed that the intrinsic subgroups can be simplified, and a set of 50 genes called PAM50 can be used to classify tumors to the four intrinsic subgroups and can be applied to predict risk of recurrence in early stage disease.
More recently The Cancer Genome Atlas Network (TCGA) performed whole exome sequencing (WES) of more than 800 primary tumors and additional high throughput analyses of subsets of these tumors. Integration of the WES and mRNA expression revealed intrinsic subtype specific mutational patterns. Overall the mutation rate was highest in basal-like and HER2-enriched tumors and lowest in luminal tumors, but significantly mutated genes were more diverse and recurrent within luminal type tumors compared with basal-like and HER2 enriched. The most common mutation in luminal A tumors are PIK3CA mutations (found in up to 45% of luminal A tumors), followed by mutations in MAP3K1 , GATA3, TP53, CDH1 , and MAP2K4. TP53 and PIK3CA mutations are the most common mutations in luminal B cancers (29% each). In basal-like tumors, the most frequent mutation was TP53 , which was found in 80% of the cases, and PIK3CA mutations were less common. In addition, loss of RB1 was found to be characteristic of basal-like tumors. Overall the basal-like cancers exhibited many molecular commonalities with high-grade serous ovarian cancers. The HER2-enriched subtype had a high rate of HER2 amplifications, and the most common mutated genes within this subgroup were TP53 and PIK3CA .
The genomic landscape in breast cancer has unveiled multiple opportunities for precision medicine, and numerous research studies are currently underway, testing targeted agents based on this information. However, clinically, off a research study, breast cancers are approached as either ER and/or PR positive, HER2 positive, or triple negative (negative for ER/PR and HER2). The most prevalent type of breast cancer is ER positive breast cancer, which is also the leading cause of breast cancer mortality.
Estrogen Receptor
ER is a transcription factor and a member of the nuclear receptor super family. ER regulates the transcription of hundreds of genes and ultimately leads to cell division, and has an important role in mammary gland development and the cell proliferation growth that occurs during pregnancy. In ER positive (ER+) breast cancers, ER has a key role in tumorigenesis, leading to uncontrolled cell division, resulting in tumor initiation and progression. Endocrine treatments that target ER are the first class of targeted treatments in cancer and are the mainstay treatment in ER+ breast cancers.
ER is composed of several conserved functional domains. These include an N-terminal transcriptional activation function (AF-1) domain; within AF-1 is a DNA-binding domain (DBD) that includes a zinc-finger domain that is responsible for recognizing the estrogen responsive element (ERE) and a second zinc finger that stabilizes protein-DNA interactions. The zinc finger domains likely have a role in tethering ER to noncanonical or imperfect ERE DNA motifs. ER has a hinge region between AF1 and the ligand binding domain (LBD), which forms the second activation domain (AF-2). The ER LBD structure is similar to those of the nuclear receptor superfamily and includes an α helical pocket, which is the site of estrogen binding as well as antagonists such as tamoxifen. After estrogen binds to ER, helices 3, 4, 5, and 12 form a groove to enable interaction with co-activators. The p160 co-activator family is the most well-characterized of ER co-activators and includes SRC1, GRIP1, and A1B1. In breast cancers, AIB1 is amplified in about 10% of tumors and overexpressed in more than 50% of tumors. Studies in engineered mouse models demonstrated the oncogenic activity of AIB1, indicating a role in breast cancer development. In addition, there are also co-repressors, such as NCOR1 and SMRT, which inhibit ER activity. ER chromatin binding is further regulated by chromatin accessibility, as many predicted EREs do not correspond to observable ER binding. The foxhead box protein A1 (FOXA1) is a pioneer factor and was found to be required for chromatin accessibility and ER binding. Furthermore, FOXA1 dictates the distribution of ER binding in a cell-dependent manner. In addition, there is also evidence of ligand independent activation of ER by receptor tyrosine kinases (RTKs), such as EGFR and HER2, which leads to distinct ER recruitment that is enriched in sites that co-localize with AP1 binding.
Endocrine Treatments
Tamoxifen
In premenopausal women, tamoxifen exerts antiestrogenic effects on the breast that are similar to those induced by surgical removal of the ovaries with resultant estrogen deprivation. The estrogen agonistic properties of tamoxifen in premenopausal women are minimal. In postmenopausal women, tamoxifen acts as an antiestrogen on breast tissue but exerts estrogen agonistic effects on the uterus, vagina, bone, pituitary, and liver. For this reason, tamoxifen is classified as a selective ER modulator, or SERM, as is its close relative, toremifene. Attempts to determine the mechanistic reasons for the divergent actions of tamoxifen on various tissues in premenopausal and postmenopausal women have led to a better understanding of the complexity of ER-mediated transcriptional regulation. Tamoxifen binds to ERα in the LBD and leads to conformational changes in the ER binding pocket at helix 12 that hinder binding of co-activators to the ER and facilitate binding of co-repressors. The continued presence of co-repressors in the complex is thought to explain the antiestrogenic properties of tamoxifen. The relative amounts of co-repressor and co-activator in certain tissues and the presence of other factors regulate whether tamoxifen acts as an agonist or antagonist. For example, upregulation of HER2 and AIB1 appear to enhance the estrogen agonistic properties of tamoxifen.
Additional estrogenic effects are mediated by membrane-initiated (extranuclear) actions at the cell membrane, as well as by tethering of the ER to binding sites on c-Jun, SP-1, IGF-R, PI-3 kinase, HER-2-Neu, c-Src.
Aromatase Inhibitors
The aromatase enzyme catalyzes the rate-limiting step in the conversion of androgens to estrogens. One steroidal aromatase inhibitor, exemestane, and two nonsteroidal, anastrozole and letrozole, are highly potent and specific. Both subclasses of inhibitor reduce aromatase to 1% to 2.5% of baseline activity in postmenopausal women, substantially reduce plasma estradiol levels, suppress tissue concentrations of this steroid in breast tumors, and are devoid of estrogen agonistic properties. Only postmenopausal patients benefit from aromatase inhibitors, since interruption of estradiol negative feedback with a reflex increase in luteinizing hormone (LH) and follicle-stimulating hormone (FSH) results in override of aromatase blockade in premenopausal women.
The efficacy of these agents in the adjuvant setting has been examined by trials comparing AIs with tamoxifen. Two similar large trials, the Anastrazole and Tamoxifen Alone and in Combination (ATAC) trial and the Breast International Group (BIG)-98 trial, compared the effects of nonsteroidal AIs (letrozole or anastrozole) versus tamoxifen with endpoints of time to progression of disease, time to treatment failure, and overall survival (OS). A third trial used the steroidal AI, exemestane. At 5 years of follow-up, all three trials demonstrated an absolute superiority of the AI of approximately 3%. The BIG-98 study reported a significant increase of 18% (hazard ratio [HR] 0.82; 95% CI, 0.70 to 0.95) in OS with letrozole, but only after correcting for selective crossover of tamoxifen patients. Albeit, improved OS was not seen in the ATAC trial. Based on these data, the AIs are the drug of choice for adjuvant treatment in postmenopausal women in the United States.
Sequential Use.
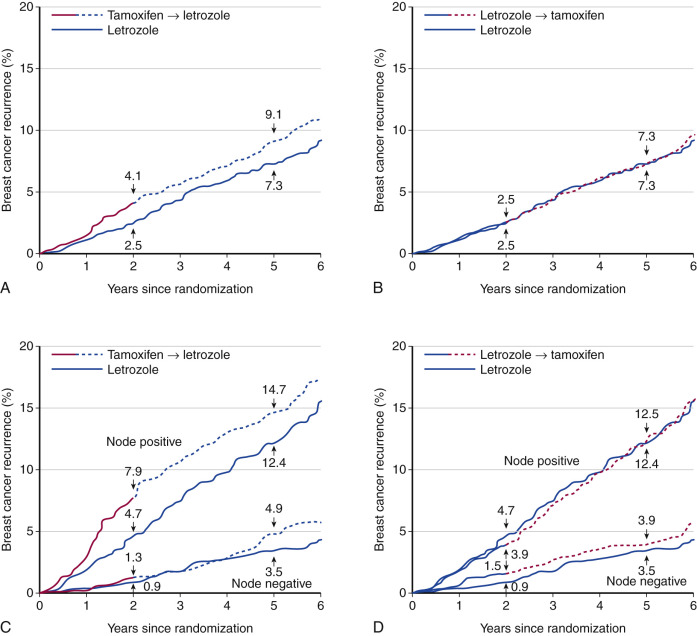
Several studies compared the sequential use of AIs or tamoxifen during adjuvant therapy. The IES study (International Exemestane Study) compared the effect of 5 years of tamoxifen with 2 to 3 years of tamoxifen with crossover to exemestane at 2 to 3 years. This study showed a reduction in new cancer events in the AI compared with the tamoxifen group. The ABCSG-ARNO trial was similar but used anastrozole rather than exemestane. The BIG-98 trial was a four-arm study comparing 5 years of tamoxifen, 5 years of letrozole, and the switching from one to the other at 2 to 3 years. The estimated disease-free percentages for the letrozole monotherapy, letrozole followed by tamoxifen, and tamoxifen followed by letrozole arms were, respectively, 87.9%, 87.6%, and 86.2%. These data suggest that either sequence is relatively equivalent ( Fig. 29.8 ) and there was no statistically significant survival benefit for letrozole versus tamoxifen. However, nonsignificant trends favor the initial use of an AI in the patients with positive nodes.
Fulvestrant.
Fulvestrant is an antiestrogen that is relatively devoid of agonist properties and therefore is called pure antiestrogen . It has two separate mechanisms of action: (1) it inhibits E2 mediated transcription by favoring binding of co-repressors to the ER complex, and (2) it increases the rate of degradation of the ER. Because of its effect to reduce the concentration of the ER, fulvestrant has now been termed an estrogen receptor down-regulator , or SERD.
The early clinical trials of fulvestrant showed comparable activity to aromatase inhibitors in advanced breast cancer after progression on first-line endocrine treatment. In addition, fulvestrant was shown to have activity in metastatic disease after progression on an AI. In a phase II prospective study in which postmenopausal patients received fulvestrant after progression on an AI, 30% of the patients derived clinical benefit at 24 weeks, and previous response to an AI did not predict response to fulvestrant. More recently, the phase II FIRST study indicated that first-line therapy with fulvestrant at 500 mg has superior activity compared with an AI. In the recent FALCON study, the 500-mg dose of fulvestrant was compared against anastrozole as first-line therapy in postmenopausal patients with metastatic, hormone receptor–positive, and HER2/ neu –negative breast cancer. Most of these patients had not received prior endocrine therapy. The FALCON data illustrated that patients treated with fulvestrant had a similar clinical benefit rate to those on anastrozole. However, progression-free survival (PFS) was significantly longer in the fulvestrant group compared with the anastrozole group (median progression-free interval = 16.6 vs. 13.8 months). Survival data were immature and additional follow-up is needed.
Resistance to Endocrine Treatment
Despite the high efficacy of endocrine treatment, resistance to endocrine treatments remains a major clinical impediment. Particularly in the metastatic setting, only approximately 50% of patients with ER+ disease will benefit from endocrine treatment, and almost all patients with initial benefit from endocrine treatment will eventually develop resistance. Many studies have focused on the investigation of endocrine resistance, and a number of underlying mechanisms are known today. These mechanisms can be categorized to four main groups, including (1) loss of ER expression, (2) ER cross talk with signaling pathways, (3) epigenetic alterations related to ER, and (4) ESR1 genomic aberrations.
Loss of ER Expression
Loss of ER expression is found in less than 20% of metastatic tumors when comparing with matched primary tumors. The etiology of loss of ER expression has not been well-studied, but putative mechanisms include either the expansion of a preexisting ER negative clone under the selective pressure of endocrine treatments or transcriptional silencing through epigenetic mechanisms. A recent study showed that the epithelial-mesenchymal transition and transcriptional repressor, Slug, inhibits the transcription of ER, supporting the latter. Genetic deletion is likely not a predominant mechanism, since recent next generation sequencing studies of metastatic breast cancers have not detected a significant number of deep deletions of ESR1.
ER Crosstalk With Signaling Pathways
A number of preclinical and clinical evidence revealed a mutual interplay between and ER and kinase signaling pathways. RTKs, such as EGFR and HER2, and downstream signaling through PI3K and MAPK can activate alternative pathways of cell proliferation and survival bypassing ER blockade. In addition, these signaling pathways can activate ER by posttranslational modifications, such as ER phosphorylation, in a ligand independent manner. Furthermore, ER can upregulate the transcription of growth factor receptors and ligands, forming a positive feedback loop. Thus this crosstalk supports the notion that in ER+ breast cancer, it is necessary to target both the ER pathway and RTK signaling pathways.
Epigenetic Alterations Related to ER
This mechanism includes changes in the ER co-regulator complexes and genome-wide ER binding sites (ER cistrome). One such mechanism is upregulation of the co-activator AIB1, which is associated with endocrine resistance in preclinical models and clinical tissue samples. Likewise, decreases in the levels of co-repressors have also been associated with endocrine resistance. In addition, changes in the FOXA1 have been linked to endocrine resistance, including a recent study that showed FOXA1 amplification in acquired endocrine resistance in a preclinical model. More globally, a number of preclinical studies and clinical tissue samples revealed that resistance to endocrine treatment can result from redistribution of ER binding, resulting in an altered transcriptional program, and this altered program can further facilitate endocrine resistance.
ESR1 Genomic Aberrations
A series of studies published recently showed that recurrent mutations in the ESR1 locus are found in about 20% of metastatic tissue samples obtained from patients that had previous endocrine treatment. These mutations detected in clinical samples were found clustered in a hotspot within the LBD. The most common mutations were mutations in the Y537 and D538 residues. Preclinical studies revealed that these are gain-of-function mutations that lead to ligand independent activation of ER. In addition, these mutations lead to relative resistance to tamoxifen and fulvestrant. The Y537 and D538 mutations reside in helix 12, a key structural part of AF2. Structural studies have shown that the Y537S ER mutation stabilizes helix 12 in the agonistic conformation, similar to wild type (WT)-ER bound to estrogen. Simulation studies and a crystallography study of the D538G mutant structure showed that this mutation also stabilizes helix 12 in the agonistic conformation in the absence of E2. In addition, the crystal structure and simulation studies of tamoxifen bound to the D538G and Y537S mutants indicate that these mutations confer an altered antagonistic conformation facilitating resistance to antagonism. Affinity studies show that these mutated receptors have a decreased affinity for tamoxifen and estrogen. Finally, several studies showed increased co-activator binding to mutant ER when compared with WT ER under ligand-independent conditions or in the presence of tamoxifen.
In addition, a number of ESR1 rearrangements have been detected, including a rearrangement that leads to a fusion protein of ER-CCD170, resulting in a truncated CCD170 protein transcribed from the constitutively active ESR1 promoter. This fusion protein is associated with luminal B type breast cancers, and in cell lines studies this protein causes endocrine resistance. Another described fusion protein is the ER-YAP1, which was found in patient-derived xenograft models. This fusion protein has loss of the ER LBD and ligand-independent activity.
New Treatments to Overcome Endocrine Resistance
Over the past five years two new classes of targeted agents have been approved for the treatment of ER+ breast cancers; these include the mTOR inhibitor, everolimus, and the CDK4/6 inhibitors palbociclib and ribociclib.
The rationale for targeting the PI3K/AKT/mTOR pathway in ER+ breast cancer is twofold. Activating PIK3CA mutations, which are common in ER+ breast cancers, are key activators of mTOR. In addition, as discussed previously, the PI3K/AKT/mTOR pathway is an important escape pathway in endocrine resistance. Indeed, everolimus, an mTOR inihibitor, in combination with exemestane, was approved for the treatment of metastatic ER+ breast cancer. This approval was based on the BOLERO study in which patients who had progressed to a nonsteroidal aromatase inhibitor were randomized to either exemestane alone or exemestane in combination with everolimus. The patients that received the combination treatment had a significantly improved progression free survival (PFS), with a median prolongation of PFS of 4.6 months.
Currently there are two CDK4/6 inhibitors that are approved for the treatment of metastatic breast cancer. The CDK4/6 inhibitor, palbociclib, was approved for metastatic ER+ breast cancer in 2015, and more recently, ribociclib, a second CDK4/6 inhibitor, was approved. CDK4/6 in complex with cyclin D1 hyperphosphorylate Rb1 enables G1-S phase cell cycle progression. Cylin D1 is a key ER gene product ; thus targeting cyclin D1 kinase activity is downstream to many of the mechanisms of endocrine resistance, such as the ESR1 LBD mutations, overexpression of AIB1, and others. Palbociclib was approved in combination with an aromatase inhibitor after the PALOMA II study in which patients were randomized to letrozole or letrozole with palbociclib as first-line treatment for ER+ metastatic disease. Patients that received the combination treatment had a significantly higher PFS (24.8 months vs. 14.5 months). More recently, the PALOMA III study was published and led to the approval of the combination of palbociclib with fulvestrant. In this study, patients who had received prior endocrine treatment were randomized to fulvestrant or fulvetrant and palbociclib, and patients on the combination arm had an improved PFS (9.5 months vs. 4.6). Ribociclib, a CDK4/6 inhibitor, was recently approved for first-line treatment of ER+ metastatic disease in combination with an aromatase inhibitor. The approval was based on the MONALEESA-2, in which patients with ER+ metastatic breast cancer without prior treatment for metastatic disease were randomized to either ribociclib with letrozole or letrozole alone. Patients on the combination arm had a significantly improved PFS (HR 0.55).
Prostate Cancer
- ◆
Prostate cancer is the third leading cause of cancer deaths in men in the United States and is primarily driven by alterations in the regulation and function of the hormone-activated androgen receptor (AR).
- ◆
Serum prostate-specific antigen (PSA) expression is used as a biomarker to assess prostate cancer development and progression and to inform about treatment options to pursue. However, since lower stage prostate cancer can be indolent and of no clinical significance, the use of PSA screening has undergone some scrutiny in more recent years.
- ◆
The advanced or metastatic, hormone-refractory stage of prostate cancer, termed castrate-resistant prostate cancer (CRPC), remains lethal and is brought about by an array of mechanisms, most of which involve alterations of the AR itself. Considerable advances have been made in available CRPC therapies, including the use of the androgen synthesis inhibitor abiraterone and the AR inhibitor enzalutamide.
Incidence
Prostate carcinoma is the third leading cause of cancer death in American men after lung cancer and colon and rectum cancer. Approximately 161,360 men will be diagnosed and 26,730 men will die from prostate cancer in the United States in 2017. The incidence of detected prostate cancer has increased exponentially over the past 2 decades, but a more gradual increase in the number of disease related deaths has been observed. Increased detection reflects not only an aging population, but also the use of more sensitive screening techniques such as the measurement of serum prostate specific antigen (PSA), first introduced in the mid-1980s. Conversely, a sharp reduction in prostate cancer incidence of more than 10% annually from 2010 to 2013 can be attributed to decreased PSA screenings following growing concerns about overdiagnosis and overtreatment. Because lower stage prostate cancer can be indolent and of no clinical significance, screening with PSA has undergone considerable scrutiny, and multiple variations have been implemented to PSA screening. These include testing of PSA density (the ratio of free to bound PSA in the bloodstream), PSA velocity, age-adjusted PSA, and PSA doubling time, the results of which have been variable. Various professional societies have therefore provided guidelines for PSA screening, both for and against, but no general agreement currently exists. However, a spike in “watchful waiting” and “active surveillance” approaches in the treatment of low-risk prostate cancer patients (defined as clinical stage T1/T2a, biopsy Gleason score of 6 or less, and a serum PSA less than 10 ng/mL) has been noted and has been correlated with an improved clinical outcome of these patients.
The global incidence of prostate cancer varies substantially; variations between countries may reflect different prevalence of risk factors, or use of screening and diagnostic methods. In Europe, the overall incidence rate was 92.1 per 100,000 in 2012, with the highest age-standardized incidence rate reported in Norway (193.2 per 100,000) and the lowest in Albania (24.8 per 100,000).
Worldwide prostate cancer is the second most common cancer in males and the fourth most common cancer overall. More than 1.1 million new cases will be diagnosed in 2012, with the highest incidence in Australia/New Zealand and the lowest in South Central Asia, although this may party reflect varying quality in data recording.
Endocrinology of Prostate Cancer Growth
Androgens
Hormonal factors, and particularly circulating androgens, play an important role in the initiation and progression of prostate cancer. Men surgically orchiectomized early in life (e.g., historically Chinese emperor-serving eunuchs or Italian castrati singers ) end up with nonpalpable or rudimentary prostates and, in theory, would rarely develop prostate cancer, although the latter has been difficult to document in the published literature. The most important advancement of our understanding of the relationship between hormones and the prostate, however, comes from the work of Charles Huggins, which eventually led to his award of the Nobel Prize in 1966. The novelty of Huggins’s findings was that the male hormones androgens (the most well-known of which is testosterone [T]) stimulated prostate tumors to grow and enhanced tumor cell survival. Huggins demonstrated that orchiectomy controlled tumor growth in men with advanced metastatic prostate cancer, at least temporarily, and prolonged their survival. Subsequent studies on Dominican Republic pseudohermaphrodites and other males who congenitally lack the type II 5-alpha reductase enzyme, an enzyme involved in androgen synthesis, helped confirm the importance of androgens in prostate growth and development. Since then, it is now fully accepted that testosterone and the 5-alpha–reduced product of testosterone, dihydrotestosterone (DHT), binds and controls the function of the AR, a hormone-regulated transcription factor that serves as a major regulator of normal prostatic and prostatic tumor growth. Approximately 7000 µg of testosterone is secreted daily by the testes, 500 µg of which is converted into DHT in various peripheral tissues ( Fig. 29.9 ); an additional 5% of total androgens is produced by the adrenal gland. These androgens include testosterone as well as preandrogens such as androstenedione, dehydroepiandrosterone (DHEA), and DHEA-sulfate, which are also converted to DHT in the peripheral tissues. In addition, a large fraction of DHT in benign and malignant prostatic tissue is produced locally in the prostate gland from circulating precursors. Therefore approximately 40% of intraprostatic DHT originates from steroids of adrenal origin.
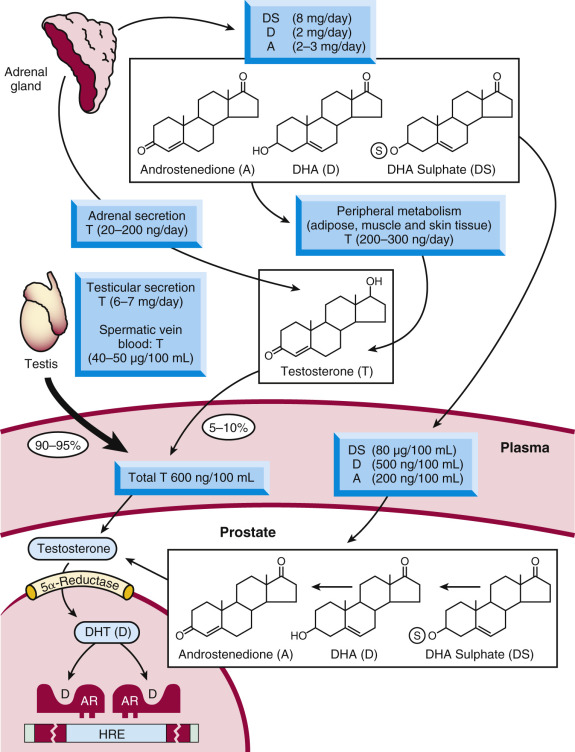
Androgen Receptor
In its non-hormone-bound state, AR is localized to the cytoplasm of target cells, in complex with molecular chaperones such as Hsp70, Hsp90, and immunophillins. In the presence of androgens, the receptor undergoes a number of confirmation changes and translocates from the cytoplasm to the nucleus, where it recognizes and binds to specific DNA sequences, known as AR response elements (ARE). Binding to AREs is facilitated by AR interactions with a number of cooperating transcription factors and coregulator proteins, and leads the expression of specific genes, some of which are involved in prostate (cancer) cell growth. The AR, like other members of the nuclear receptor family, consists of an N-terminal domain, a highly conserved DBD, a carboxy-terminal LBD, and a hinge region (which joins the LBD to the DBD). For most nuclear receptors, transactivation occurs through a region in the LBD termed activation function 2 (AF2) that facilitates ligand-dependent transcriptional activation. The LBD consists of 12 helical structures, and upon hormone binding, the 12th helix is reorganized to an agonist conformation, generating a hydrophobic surface termed the “coactivator pocket” for tissue-specific coactivator binding. Coactivators bind to this hormone-induced conformation via a conserved “LXXLL” sequence to enhance the transcriptional activity of the receptor. All clinically approved AR targeting antiandrogens for prostate cancer therapy (described in more detail later in the chapter) function by competing with the endogenous androgens for the ligand-binding pocket of the AR. Paradoxically, most of the transactivation function of the receptor lies at the AR N-terminus, subdivided into transactivation units (τ1: amino acids 100 to 360) and (τ5: amino acids 360 to 528). Although targeting the AR N-terminus has been historically challenging, due to a lack of enzymatic activity or rigid binding clefts in that domain, recent therapeutic advances have been made.
Mechanisms of Endocrine Therapy Resistance
Despite the effectiveness of endocrine therapies in prostate cancer, intrinsic and acquired resistance remains a clinical challenge. Typically, after an initial response to antiandrogen treatment, tumors will progress, and this will usually be in concert with an increase in serum PSA. Secondary responses to blockade of adrenal androgens may occur, as evidenced by a 50% or greater decrease in PSA levels and often by pain reduction and improvement of fatigue, but rarely by objective regression of tumor mass or healing of bone lesions. This stage of hormone ablation unresponsive prostate carcinoma remains incurable to date and is referred to as CRPC. CRPC is brought about by an array of mechanisms, most of which involve alterations of the AR itself. For example, AR may develop hypersensitivity to residual androgens, or acquire the ability to become activated in a ligand-independent manner through mutations or alterations of key phosphorylation sites on the receptor. More recently, the occurrence of AR variants that lack some or most of the regulatory domains of the full-length AR have been observed. These AR variants are constitutively active and do not respond to antiandrogen treatment. Their significance remains unclear, although they have been linked to resistance to androgen ablation therapy. In addition, it is yet to be fully understood how and why most of these variants are expressed; hypotheses range from alterations in splicing factor expression to select amplifications or rearrangements of the AR gene locus itself.
A number of growth factor pathways are shown to be upregulated in prostate cancer and also lead to androgen-independent activation of AR. For example, the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway regulates activity of the AR through phosphorylation of the receptor itself, as well as downstream targets, resulting in resistance to apoptosis and increased cell survival. In addition, mutations or inactivation of PTEN are commonly found in advanced prostate cancer. PTEN is a phosphatase that dephosphorylates and inactivates PI3K. With mutational inactivation of PTEN, an associated upregulation of phosphorylated, activated AKT occurs, as well as downstream activation of mTOR. This in turns leads to significant changes in cell proliferation. Other common genetic alterations in prostate cancer include losses of NKX3.1 and recurrent mutations in MED12 , FOXA1 , and SPOP , although the exact mechanisms by which these lead to prostate cancer progression remain to be fully understood.
Etiology and Risk Factors
Primary nonmodifiable risk factors for prostate cancer development include age, race, and family history. The SEER registry reports that the incidence of prostate cancer increases with age from 9.2 per 100,000 between ages 40 and 44 years old to 984.8 per 100,000 at ages 70 to 74 years old. Incidence is increased in African American males with an age-adjusted RR of 1.73 (95% CI, 1.23 to 2.45). Their mortality rate is nearly double compared with that of Caucasian males. Genetic factors are also important; the RR for a man with one first-degree relative with prostate cancer increases more than twofold, with even higher risk with multiple first-degree relatives. Overall, men with prostate cancer report a positive family history 3.1 to 4.3 times more commonly than men without prostate cancer, and twin studies have shown a genetic component in 42% (95% CI, 0.29 to 0.50).
Genetic Alterations
Recent genome-wide association studies have identified 46 susceptibility loci in prostate cancer. Candidate genes based on linkage analysis include BRCA2 . BRCA2, but not BRCA1, is also associated with prostate cancer in Ashkenazi Jews, whereas most series in unselected populations do not show this association. Mutations and alterations of the AR itself are also commonly observed. Although the AR mutation rate in early stage prostate cancer is quite low, it significantly increases (by up to 40%) as the cancer progresses. The frequency and type of mutation appears to be influenced by selective pressure—namely that exerted by antiandrogen treatment. For example, men treated with flutamide exhibit higher levels of mutations (primarily at residue 877) compared with men not receiving antiandrogens. Several epidemiological studies have also reported a correlation between the number of polyglutamine repeats (CAG) on the AR and the incidence of prostate cancer, but others were not able to confirm this finding.
Other candidate genes implicated in prostate cancer development include the metal dependent hydrolase ELAC/HPC2 , RNASEL (an endo-ribo-nuclease), macrophage scavenger receptor 1 gene (MSR1) , CYP17A1 (cytochrome p450 enzyme responsible for the 17 hydroxylation of steroids), and SRD5A2 (the predominant form of 5-alpha reductase in the prostate). Moreover, fusions between ETS family transcription factor genes (e.g., EGR and ETV1 ) and TMPRSS2 are commonly found in prostate cancer (i.e., in more than 90% of the 57% of tumors that overexpress EGR or fusion between TMPRSS2 and ETS gene [ETV1] ) and may play an etiological role.
Modifiable Risk Factors
Modifiable risk factors that have been proposed include diet, BMI, smoking, and physical activity. The significant differences in age standardized global incidence rates provide highly suggestive evidence that dietary and environmental factors play major roles in risk. Differential ingestion of high fat diets, red meat, green tea, or soy products provide potential explanations for the divergent rates of prostate cancer among different populations. Several case-control studies examining Western diets have shown a positive association with prostate cancer risk, but these studies are confounded by potential recall bias and do not prove causality. More recently, a correlation between diet and changes in the prostate cancer epigenome have been reported. Others suggest that components of vegetables such as lycopene or antioxidants such as vitamin E and selenium may reduce the risk of prostate cancer. However, results examining dietary supplements have been mixed, possibly in part due to the difficulty in accurately recording the true intake of vitamins and minerals. Surprisingly, studies including the Cancer Prevention Study and the National Institutes of Health-American Association of Retired Persons Diet and Health Study demonstrate that multivitamin users were at higher risk for fatal prostate cancer compared with those men who did not consume multivitamins. One study, the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study, which supplemented alpha-tocopherol (50 mg/day) to elderly Finnish smokers, did demonstrate a decreased risk of prostate cancer. However, the Selenium and Vitamin E Cancer Prevention Trial, enrolling more than 35,000 men, did not find an association between prostate cancer risk and either vitamin E or selenium. In summary, prospective studies of dietary factors and supplements have not confirmed beneficial effects of any of these components.
Inflammation
Chronic or recurrent inflammation may play a role in the development of prostate cancer. Prostatitis, diagnosed by symptoms, occurs in 9% of men between 40 and 79 years old. Inflammatory cells release oxidants that could result in DNA damage and act as initiators of prostate cancer. A lesion called “proliferative inflammatory atrophy” has been proposed as a precursor to prostatic intraepithelial neoplasia (PIN) and prostate cancer. Chronic inflammation is associated with proliferative inflammatory atrophy and signs of molecular stress such as high levels of glutathione-S-transferase A1 and cyclooxygenase-2 (COX-2). These observations suggest that inflammation increases oxidative stress, which in turn causes DNA adducts that could then contribute to prostate cancer development.
Prevention
A large randomized, controlled trial, the Prostate Cancer Prevention Trial (PCPT), tested the hypothesis that reduction of DHT formation with finasteride, an inhibitor of type II 5-alpha reductase, would reduce prostate cancer incidence. The trial, initiated in 1993 and enrolling 18,882 men aged 55 or older with a normal digital rectal exam (DRE) and initial PSA of 3.0 ng/mL or lower, included 9060 men in the final analysis. A biopsy was performed at the end of the study (7 years) or for clinical indications (e.g., abnormal DRE or adjusted PSA of 4.0 ng/mL or higher). At the end of 7 years of therapy, 4368 men receiving finasteride and 4692 receiving placebo were available for analysis. The finasteride group experienced a 24.8% reduction in prostate cancer diagnosis (95% CI, 18.6 to 30.6; P < .001). However, tumors of Gleason grade 7 to 10 occurred in 15% more men receiving finasteride (280 of 757 tumors or 37%) than in those taking placebo (237 of 1068 tumors or 22%; P < 0.001). High-grade disease occurred in 6.4% of the finasteride group and 5.1% of the placebo group. Five deaths due to prostate cancer occurred in each study arm. Side effects in the finasteride group included reduced volume of ejaculate, erectile dysfunction, loss of libido, and gynecomastia ( P < .001 vs. placebo), but attributable risks of these side effects ranged only from 1% to 13%. These results stimulated multiple follow-up studies to identify possible reasons for the finding of an increase in higher grade lesions. Possible explanations included pathological artifact, volume-grade artifact, or detection bias. One of the effects of finasteride is shrinkage of normal prostate volume to 50% of the original size. Therefore it is theoretically possible that biopsy of larger prostates (i.e., men on placebo) would result in fewer high-grade cancers detected and that the true incidence would be similar between the two groups. Notably, this detection bias was demonstrated in retrospective reviews. The results could be due to selection of androgen-independent clones induced by lower intraprostatic DHT, as has been observed in hypogonadal men with prostate cancer. Polymorphisms of genes involved in estrogen metabolism that are associated with increased risk of prostate cancer could have been involved. Since the Gleason grading system was created based on prostate tumors that were hormonally naïve, finasteride may have altered the diagnosis of high-grade cancer by modulating the influence of the epithelium. Despite the evidence that finasteride did decrease the incidence of low-grade prostate cancers, debate continues as to whether it should be used in prostate cancer prevention. Analysis of the lifetime implications, side effects (e.g., erectile dysfunction, loss of libido, and gynecomastia), and cost-effectiveness of using finasteride for prostate cancer prevention does not appear to support its routine use.
The Reduction by Dutasteride of Prostate Cancer Events (REDUCE) Trial examined the possibility that blocking both type I and type II 5-alpha reductase might be superior to blocking only type II. Dutasteride, approved in 2002, blocks both type I and type II 5-alpha reductase subtypes and putatively might be superior for prevention of prostate cancer. Dutasteride, in comparison with finasteride, induces a greater reduction in both serum (94.7% vs. 70.8%) and intraprostatic (greater than 90% vs. 68% to 86%) DHT levels. The REDUCE trial enrolled a total of 6729 men with the following characteristics: ages 50 to 75 years old, initial PSA of 2.5 to 10 ng/mL, and negative biopsies at the time of enrollment. Among the men randomized to 0.5 mg/day of dutasteride ( n = 3305) or placebo ( n = 3424), 659 and 858 developed prostate cancer within the 4-year study period, demonstrating a 22.8% (95% CI, 0.152 to 0.298) reduction in RR of prostate cancer in the treated group. Unlike the PCPT trial, however, no increased risk of high-grade cancers was noted with dutasteride. Although the lack of an increase in high-grade cancer in the REDUCE trial would support that this finding was an artifact in the PCPT, these studies were designed differently. Most importantly, subjects in the REDUCE trial underwent a biopsy at the beginning of the study and were enrolled only if this was negative. Therefore, in the PCPT, some subjects may have harbored occult prostate cancer that went undetected until well into the study.
Epidemiological and secondary endpoint data suggest that other approaches, such as supplementation of selenium, vitamin E, and lycopene, might prevent prostate cancer, but available data are not supportive. The Selenium and Vitamin E Cancer Prevention Trial (SELECT) randomized 34,887 men aged 55 years old or older (50 years old if African American) with normal DRE and PSA of 4.0 ng/mL or less, to receive selenium, vitamin E, both agents, or placebo. No significant differences in prostate cancer incidence were observed, and the study was stopped in 2008. Notably, extended follow-up through 2011 demonstrated an actual increased risk in prostate cancer development with vitamin E.
Treatment Strategies in Localized Disease
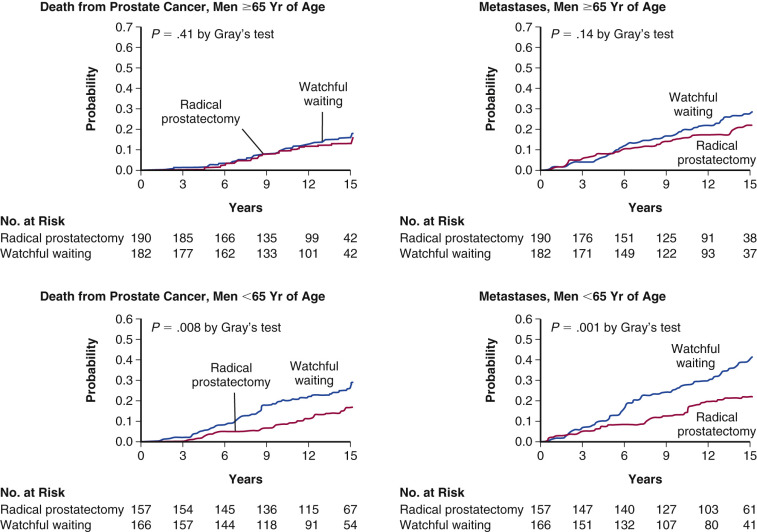
Three competing strategies are recommended for management of localized prostate cancer: active surveillance, radical prostatectomy, and radiotherapy. A key question is whether radical prostatectomy prolongs disease-free survival (DFS) and OS when compared with surveillance. This issue has recently been examined in a randomized, controlled, clinical trial comparing radical prostatectomy with surveillance in men with clinically localized disease. Six hundred and ninety-five men with localized prostate cancer, a life expectancy of greater than 10 years, and a mean age of 64.7 years were randomized and followed for a median of 8.2 years ( Fig. 29.10 ). At the time of the initial study, 83 men in the surgery group and 106 in the surveillance group died of all causes ( P = .04). Death was due to prostate cancer in 8.6% of the surgical group and 14.5% of the surveillance group. The difference in death rates became greater at the 10-year time period (5.3 percentage points) than at the 5-year time frame (2.0 percentage points). The risk of distant metastases was reduced to 1.7% from 10.2% by surgery and the rate of local progression from 25.1% to 19.1%. Further follow-up in 2008 and 2011 confirmed the reduction in prostate cancer deaths in patients undergoing surgery. Although this study does demonstrate improvement in both OS and DFS with surgery, the effect is rather small. Nonetheless, the decrease in incidence of metastases and local tumor progression can be considered clinically significant.
Observational studies over several decades demonstrated the efficacy of radiotherapy (both delivered in the conventional external beam format and as brachytherapy) as definitive therapy in men with prostate cancer and as an alternative to surgical prostatectomy. Recent studies addressed whether neoadjuvant or adjuvant androgen deprivation therapy enhances the overall efficacy of therapy in men with aggressive disease. The Radiation Therapy Oncology Group (RTOG) Trial 86-10 examined the role of neoadjuvant endocrine therapy prior to radiation. The incidence of local progression at 5 years was 46% for patients with T2-4 tumors receiving adjuvant hormonal therapy and 71% for those receiving radiation alone ( P = .001), but no difference in OS was noted. Studies examining neoadjuvant hormonal therapy prior to prostatectomy demonstrate no change in cause-specific survival but do show lower risk of positive margins at the time of surgery. Current clinical trials examining neoadjuvant abiraterone, an irreversible inhibitor of 17α-hydroxylase/C17, 20 lyase (CYP17A1) that is approved for the management of CRPC, are currently underway.
Radiotherapy with concomitant (adjuvant) androgen-deprivation therapy has been the subject of two randomized trials. A RTOG study involved patients with T3 and T4 disease given radiation therapy followed by medical castration with goserelin starting at the last week of irradiation. DFS at 5 years was 60% for the goserelin arm and 44% for radiation alone ( P < .0001). Only the subset of men with Gleason grade 8 to 10 tumors experienced a survival benefit from 66% to 55% ( P = .03). However, the 10-year follow-up demonstrated a survival benefit for all patients treated with adjuvant hormonal therapy on multivariate analysis (HR, 1.3; P < .001). DFS was also improved (HR, 2.2; P = .0003). In a similar EORTC trial, the 5-year DFS was 85% in the adjuvant hormone treated group (goserelin) versus 48% in the radiation alone group ( P = .001). Importantly, OS was significantly increased in the adjuvant hormonal group (79% vs. 62%; P = .001).
The majority of data favor the use of adjuvant hormonal therapy in patients undergoing radiation therapy as definitive treatment for prostate cancer, but only for high-risk localized or locally advanced disease. Analysis of pooled data from the RTOG neoadjuvant and adjuvant trials concluded that the key consideration may be the necessity for long-term rather than short-term hormonal therapy in either neoadjuvant or adjuvant therapy approaches. In those men receiving short-term hormonal therapy (goserelin and flutamide for 2 months before and 2 months after radiotherapy), statistically significant improvements were observed for the endpoints of distant metastasis-free and no evaluable disease (NED)–free intervals but not for OS. Only long-term adjuvant therapy improved OS, and only in the subset of men with Gleason grade 7 to 10 tumors. These conclusions on long-term versus short-term adjuvant hormonal therapy were further supported by a nonrandomized Canadian trial.
Use of antiandrogen therapy is not indicated for clinically localized disease. The Early Prostate Cancer (EPC) program examined the use of 150 mg of bicalutamide in addition to standard treatment (e.g., radiation, prostatectomy, or surveillance) for clinically localized disease (71% of 2285 patients) or locally advanced but nonmetastatic disease (29%). At a median follow-up time of 9.7 years, PFS was significantly improved in the bicalutamide group (HR 0.85; P = .001), although there was no significant improvement in OS. Patients with locally advanced disease treated with radiotherapy derived the greatest benefit in terms of OS. However, patients with localized disease did not benefit in terms of either PFS or OS.
Clinical Therapies in Locally Advanced or Metastatic Disease
Androgen deprivation represents the earliest form of systemic therapy for locally advanced and metastatic prostate cancer, and observational studies have demonstrated its efficacy. Three methods have been historically used: surgical castration, high-dose estrogen, and GnRH agonist analogues. Surgical castration as well as high-dose estrogen in the form of diethylstilbestrol (DES) has been used as treatment of prostatic cancer since the 1940s. In the VACURG studies, 5 mg of DES decreased the rate of recurrence of prostate cancer but increased the cardiovascular death rate. Following this observation, careful dose response studies indicated that 3 mg of DES daily minimizes the risk of cardiovascular disease acceleration and maximizes the beneficial effects on prostate cancer. Gynecomastia and impotence are the major side effects. However, DES is no longer available in the United States.
GnRH superagonist or antagonist analogues are now available, which suppress testosterone to castrate levels and cause tumor responses equivalent to those induced by castration. Comparison of the various monotherapies was the subject of a meta-analysis involving 10 separate trials which concluded that orchiectomy, GnRH agonist analogues, and DES produced equivalent survival in men, and no differences existed among the various GnRH analogues (GnRH-A) currently available. The decision between surgical castration and GnRH-A represents a major question. Surgical orchiectomy produces a rapid decrease in serum androgen levels; it does not require patient compliance long-term and is effective in inducing tumor regression in nearly 90% of patients. However, more than half of men in the United States prefer medical castration as a means to avoid surgery, most likely due to the psychological impact of castration.
Highly potent agonist analogues of GnRH have been approved for use in prostate cancer and effectively produce a “medical orchiectomy.” These compounds paradoxically inhibit LH secretion by the pituitary suppressing testicular testosterone production. Initially, GnRH-A administration increases LH threefold to fourfold and testosterone twofold for 1 to 2 weeks. Thereafter LH is profoundly suppressed and plasma testosterone levels fall from approximately 500 ng/dL to castrate levels of 15 ng/dL. Hormonal inhibition continues for up to 2 years of continuous therapy. The initial rise in testosterone causes a transient disease flare in 5% to 10% of patients, producing an objective increase in tumor size in approximately 3% of patients, and a subjective increase in bone pain from metastases in the remainder. Although tumor flare is usually transient, severe reactions with spinal cord compression or death have been observed in the occasional patient. Based on this, it is necessary to administer an antiandrogen such as bicalutamide during the initial few weeks of GnRH-A therapy.
Newer GnRH receptor antagonists have been developed to block the interaction of GnRH with its receptor on the pituitary gland, resulting in an immediate and substantial reduction in LH and testosterone levels without causing the flare phenomenon. In a phase III, randomized clinical trial, degarelix, approved by the FDA in 2008 for patients with advanced prostate cancer, demonstrated equal effectiveness compared with leuprolide at suppressing testosterone to castrate levels. Notably, degarelix suppressed levels of both testosterone and PSA significantly faster, and no patient experienced a testosterone surge compared with 81% of leuprolide-treated patients.
The major rationale for using a GnRH-A is to induce castrate testosterone levels without requiring surgical removal of the testes. Both testosterone and DHT levels in patients treated with either a GnRH-A or orchiectomy fall to a similar extent. Objective regressions occur as frequently as with orchiectomy, and a meta-analysis study including 10 randomized controlled trials with 1908 patients provided evidence that medical and surgical castration exert equal clinical effects. Initially, a major problem with GnRH-A therapy was the requirement for daily subcutaneous administration, with the possibility of noncompliance with daily injections and incomplete androgen suppression. Third-generation formulations are now available, which allow injections at 3- to 6-month intervals or implants yearly. These biodegradable preparations appear highly effective, well tolerated, and acceptable to patients. Because some androgen responsive cells remain in prostate tumors after relapse on the GnRH agonists, continued GnRH-A treatment is advocated upon disease relapse and preferably for life.
Hormonal Therapy for Locally Advanced or Metastatic Disease
Historically, approximately 25% of men presented with either metastatic or lymph node positive disease. However, currently less than 5% of men present with synchronous metastatic disease. Whether to initiate hormonal treatment immediately in these patients or to defer until symptomatic is a major question, especially considering recent evidence demonstrating increased cardiovascular disease and mortality with use of androgen deprivation therapy. Because cure with hormones is not possible, the two goals of treatment are to increase life expectancy and to relieve symptoms. If treatment were to be advocated in asymptomatic patients, the therapy should improve the length of patient survival. Rigorously controlled series from the Veterans Administration Cooperative Urological Research Group (VACURG) trials during the 1960s indicated no clear survival benefit from endocrine therapy in patients with stage C or D disease (i.e., advanced) at the time of diagnosis.
Based upon these findings, standard practice usually involved withholding hormonal therapy until symptomatic metastatic disease developed. However, two studies provide evidence favoring therapy immediately upon detection of metastatic disease. Messing and colleagues studied men with prostate cancer found to have positive pelvic lymph nodes at the time of radical prostatectomy. The study randomized men into either an immediate therapy group (medical or surgical orchiectomy) or into a group observed until an indication for hormonal treatment occurred. Death due to prostate cancer occurred in 3/47 men in the immediate treatment group and 16/51 in the deferred group ( P < .01). At a median follow-up of 11.9 years in 2006, there was continued benefit of early androgen deprivation in OS, DFS, and CSS. In another study, 934 men with locally advanced prostate cancer or with asymptomatic metastatic disease were randomized to immediate versus deferred hormonal therapy. An initial report concluded that there was an OS benefit from immediate therapy. Overall, 203 men died from prostate cancer in the immediate therapy arm and 257 in the deferred group ( P = .02). However, on extended follow-up, the OS benefit was no longer statistically significant. Regardless, deaths due to prostate cancer were reduced to 241 in the immediate therapy arm versus 287 in the deferred arm ( P = .0019). In this group of elderly men, deaths from other causes occurred commonly and were not influenced by immediate hormonal therapy. Nonetheless, there were fewer cases of cord compression (10 vs. 24), pathological fracture (14 vs. 22), extraskeletal metastases (47 vs. 62), and ureteral obstruction (49 vs. 64) in the immediate treatment group.
Another study randomized men not accepting radical prostatectomy to immediate or deferred androgen therapy and found no differences in overall pain-free time and performance status. CFS tended to be longer in the immediate group ( P = .09) but OS was identical ( P = .96). Other evidence of efficacy of early therapy comes from nonrandomized studies from the Mayo Clinic that demonstrated a statistically significant survival advantage for early endocrine therapy (castration) in men with stage D1 (T0-3, N1-2, M0) disease. Seventy-three patients underwent either radical prostatectomy or radical prostatectomy combined with orchiectomy. An advantage for immediate adjuvant therapy was demonstrated as 5-year survival rates were 93% in the immediate orchiectomy group and 80% in the deferred orchiectomy group. In another study, benefit was seen only in patients with diploid and not in those with tetraploid or aneuploid tumors. Finally, a subset analysis of men in the RTOG trial number 85-31 study examined a group of 139 men with capsular penetration or seminal vesicle involvement; 71 men received RT plus LH-RH agonist therapy and 68 men received RT alone. Statistically significant improvement in PFS and freedom from biochemical relapse (i.e., PSA) was observed in the LH-RH group. However, there was no statistically significant difference in OS in this relatively small subset of men followed for a median of 5 years only.
A systematic review in 2002 concluded that early androgen deprivation therapy resulted in significant reductions in both DFS and complications due to progression. PFS was significantly higher at years 1, 2, 5, and 10 (odds ratio [OR], 3.99, 4.79, 3.15, and 3.49, respectively) in men treated with early androgen deprivation, and OS at 10 years was also improved (OR, 1.5; 95% CI, 1.04 to 2.16). Taken together, these data suggest that immediate hormonal therapy may be efficacious for asymptomatic men presenting with lymph node spread. Nonetheless, in elderly men with increased risk of dying of nonprostate cancer causes, deferred therapy appears to be a reasonable choice.
The American Society of Clinical Oncology (ASCO) critically analyzed these data and commented that all trials cited previously were conducted before the routine use of PSA testing. The ASCO review was considered to be limited by the variability in the interventions as well as in the stages of the cancers of the patients enrolled. From this analysis, the guidelines concluded that current data do not provide definitive evidence that early endocrine therapy is beneficial regarding improved survival of patients presenting with metastatic disease. Longer follow-up and additional studies sufficiently powered to detect an increase in OS are needed before definitive conclusions are warranted.
Men who experience a rise in PSA after an initial fall to undetectable levels after radical prostatectomy might also benefit from hormonal therapy. Data indicate that a rapid rise in PSA suggests the presence of metastatic disease, whereas a slow rise suggests local recurrence. In a single small study, 68% of men progressed to detectable clinical disease upon observation for a median of 19 months. With adjuvant hormonal therapy or radiotherapy, the rate of progression to clinically detectable disease decreased to 21%.
Medical or surgical castration causes an abrupt reduction of androgen levels resulting in hot flashes in up to 80% of patients, 27% of whom report this to be the most troublesome adverse event. Bone density falls in men in response to declines in androgens and their aromatized estrogen metabolites, and the risk of fracture increases starting 1 year after initiation of therapy. Either pamidronate or zoledronic acid, both potent intravenous bisphosphonates, can completely abrogate the effects of androgen deprivation on bone. Significant erectile dysfunction occurred in 78.6% and 73.3% of men after orchiectomy and GnRH-A therapy, respectively. Other metabolic effects that occur as a result of androgen deprivation therapy include a decrease in hemoglobin and lean body mass, increase in total body fat mass, and rises in total cholesterol and triglyceride levels. Fatigue and psychological distress tend to be more common in men receiving active therapy compared with those choosing to defer treatment. Finally, gynecomastia occurs in 1% to 16% of men treated with androgen deprivation therapy.
Secondary Hormonal Therapies for Recurrent Disease
Disease that recurs following medical or surgical castration can be treated with secondary hormonal therapies. Prior to the PSA era, the clinician had to rely on bone x-rays and soft tissue changes to document responses, and these were rarely observed. Consequently, the efficacy of secondary hormonal therapies was controversial. More recently, clinicians have accepted a 50% decline in PSA as reflecting an objective response to therapy. Studies demonstrate that this PSA endpoint predicts a significantly prolonged median OS, objective PFS, and time to pain progression. Using PSA measurements, it has now been possible to show efficacy of a number of secondary hormonal therapies. Responses to antiandrogens, such as ketoconazole, abiraterone, enzalutamide, flutamide, bicalutamide, aminoglutethimide, DES, and glucocorticoids alone, range from 14% to 60%. Head to head comparisons are unavailable for these agents, and relative efficacy cannot be compared ( Table 29.1 ).
Related posts:
 Gonadotropin Hormones and Their Receptors
Gonadotropin Hormones and Their Receptors
 Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
 Structure, Function, and Evaluation of the Female Reproductive Tract
Structure, Function, and Evaluation of the Female Reproductive Tract
 Endocrine Disturbances Affecting Reproduction
Endocrine Disturbances Affecting Reproduction
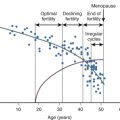 Medical Approaches to Ovarian Stimulation for Infertility
Medical Approaches to Ovarian Stimulation for Infertility
 Pelvic Imaging in Reproductive Endocrinology
Pelvic Imaging in Reproductive Endocrinology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


