Hormonal Therapy of Breast Cancer
Beverly Moy
Paul E. Goss
Hormonal therapy plays a critical role in the prevention and treatment of breast cancer at all stages of its pathogenesis.1 This chapter provides an understanding of (a) estrogen synthesis and metabolism, (b) the endocrine regulation of these processes, (c) the molecular basis of estrogen signaling through the estrogen receptor (ER), (d) the response of cancer cells to the modulation of ER signaling, (e) the pharmacology of hormonal therapeutics, and (f) hormonal therapy resistance.
Estrogen Structure, Biosynthesis, Transport, and Metabolism
Structure of Estrogen
Estrogens are steroids containing a hydrated four-ring structure (cyclopentane-perhydro-phenanthrene), in which a five-sided cyclopentane ring (designated the D ring) is attached to three six-sided phenanthrene rings (designated the A, B, and C rings) (Figs. 35-1 and 35-2).
The three endogenous estrogens are estradiol, estrone, and estriol. Estradiol and estrone can be formed directly by aromatization of testosterone and androstenedione, respectively. Estradiol can also be readily formed from estrone and is the principally active estrogen (Fig. 35-3).2, 3, 4, 5 The common and systematic names of selected steroidal hormones and their structures are shown in Figure 35-2.2, 3, 4, 5 The physiologic effects of estrogens are summarized in Table 35-1.
Estrogen Biosynthesis
Estrogen biosynthesis begins from cholesterol. Cytochrome P-450 enzymes convert cholesterol to different steroid hormones via alteration of side chains on the molecule.6 The final step in estrogen synthesis is aromatization, which is catalyzed by the P-450 aromatase monooxygenase enzyme complex.6,7 The hormonal regulation of the CYP19 gene on chromosome 15, which spans 120 kb, is intricate, primarily due to a complex and tissue specific promoter structure, with regulatory elements targeted by gonadotropins, glucocorticoids, growth factors, cytokines, and the intracellular signaling molecule cAMP.8, 9, 10, 11
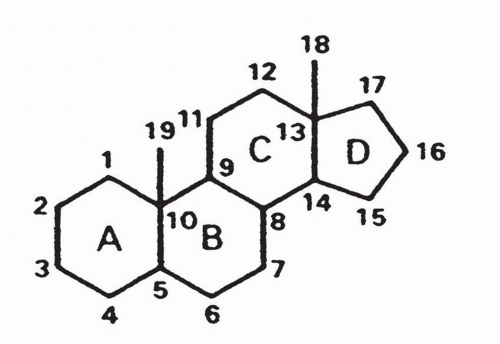 FIGURE 35-1 The structure and numbering of the cyclopentane-perhydro-phenanthrene nucleus. |
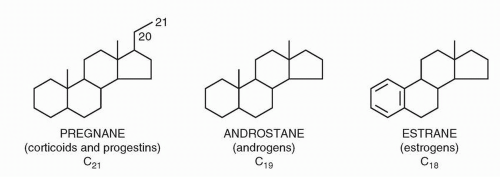 FIGURE 35-2 The structure and names of the five major steroid hormones. (Trivial name is followed by systematic.) Cortisol, 4-pregnen-11β,17α, 21-triol-3,20-dione; aldosterone, 4-pregnen-11β,21-diol-18-al-3,20-dione; progesterone, 4-pregnen-3,20-dione; testosterone, 4-androsten-17β-ol-3-one; estradiol, 1,3,5(10)-estratrien-3,17β-diol. |
In premenopausal women, estrogens are predominantly produced by the ovaries. By contrast, in postmenopausal women, estrogens are instead formed from the extragonadal conversion of ovarian and adrenal androgens via aromatase. Androgens from the ovaries and adrenal glands are released into circulation and converted to estrogens in tissues with aromatase activity, including adipose tissue and muscle.12
Estrogen Transport
Estrogens circulate in the bloodstream predominantly bound to albumin and steroid-binding globulins. The estrogens and androgens are transported via testosterone and estradiol-binding globulin or sex steroid-binding globulin.13 The unbound, or free, hormone, which makes up only 2% to 3% of total circulating estrogens, enters the cell by diffusion through the cell membrane.6 Once inside the cell, estrogens bind to the ER, thereby starting a complex series of signaling events in the target cell. Local estrogen synthesis within tissues such as the breast may also act as a paracrine factor.14
The Estrogen Receptor
ER Structure
The ER is a member of the nuclear hormone receptor superfamily that includes the progesterone receptor (PgR), androgen receptor (AR), glucocorticoid receptor (GR), and mineralocorticoid
receptor. This receptor family also includes receptors for nonsteroidal nuclear hormones such as the retinoids, vitamin D or deltanoids, and thyroid hormones.
receptor. This receptor family also includes receptors for nonsteroidal nuclear hormones such as the retinoids, vitamin D or deltanoids, and thyroid hormones.
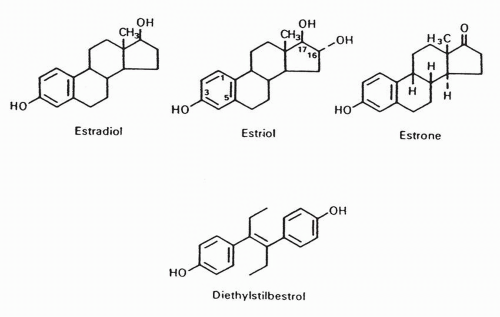 FIGURE 35-3 Endogenous and synthetic estrogens. |
The ER, like most nuclear hormone receptors, operates as a ligand-dependent transcription factor that binds to DNA at estrogen response elements (EREs) to direct changes in gene expression in response to hormone binding.6,15 The ER protein structure includes six domains, designated A to E (Fig. 35-4). Estradiol binds to the ligand-binding site in the E domain. The E domain also mediates ER dimerization, with assistance from residues in domain C. The sequence-specific DNA-binding function resides in domain C. Domain D contains a nuclear localization signal required for transfer of the ER from the cytoplasm to the nucleus. Sequences that promote transcription, or activation functions (AFs), are present in domains A and B (AF1) and domain E (AF2). The basic structure and functional components of steroid hormones follow the same pattern, with a hormone-binding site, a dimerization domain, transactivation domain(s), and a nuclear localization signal.16
ER Subtypes: ERα and ERβ
The first identified ER, now known as “ERα,” was discovered in the late 1960s, and the gene was cloned in 1986. The second ER, named “ERβ,” was first reported in 199617 and has provided a further layer of complexity to our understanding of estrogenregulated gene expression.18 Furthermore, each subtype of ER exists in several isoforms. At the amino acid level, ERα and ERβ are highly homologous in the DNA-binding domain (96%), but the homology in the ligand-binding domain (LBD) is only 58%. This structural comparison suggests that the two subtypes would recognize and bind to similar DNA sites. However, the differences in the LBD suggest that responses to different ligands may be more distinct than anticipated from primary sequence analysis.19
TABLE 35.1 Physiologic effects of estrogens | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
There is no consensus on the clinical significance of ERβ since many different studies have conflicting findings. However, breast cancers that coexpress ERα and ERβ tend to be node-positive and of a higher grade than tumors that express ERα alone20 ERβexpressing tumors tend to be PgR-negative.21 These correlations suggest an adverse effect of ERβ expression on prognosis. Other work, on the other hand, suggests that decreased levels of ERβ may be associated with more aggressive clinical behavior.18
Classical ER Signal Transduction
Ligand-Dependent ER Signaling
The well-described classical ER signaling pathway is illustrated in Figure 35-5. Estrogens bind to the LBD of the ER, leading to the release of the receptor from heat shock protein 90. This ligand binding is then followed by phosphorylation of the receptor at specific serine residues, ER dimerization, and then sequence-specific DNA binding to a sequence referred to as an “estrogen response element”. In the presence of estrogen, messenger RNA (mRNA) transcription is promoted through AF2. Residues in AF1 also promote transcription, although the function of AF1 does not require the presence of estrogen.22 The consensus DNA-binding sites for each of the nuclear hormones are illustrated in Figure 35-6. While the ER binds most strongly to the ERE consensus sequence, it is also capable of promoting transcription through sequences that have only partial homology to a classic ERE. In these cases, nearby response elements for other transcription factors (e.g., SP-1) contribute to ER activity.22, 23, 24 The characteristics of the target gene promoter are critical to the specific nuclear actions of the activated ER. Other factors that are critical are the structure of the
bound ligand and the balance of coactivators and corepressors associated with the ER-ligand complex. In addition, ERα and ERβ can either homodimerize or heterodimerize, and this has an impact on their activity at the DNA-binding site.25
bound ligand and the balance of coactivators and corepressors associated with the ER-ligand complex. In addition, ERα and ERβ can either homodimerize or heterodimerize, and this has an impact on their activity at the DNA-binding site.25
 FIGURE 35-4 Examples of amino acid homology between nuclear hormone receptors. A/B, activation function 1 transactivation domain; C, DNA-binding and homodimerization domain; D, nuclear localization signal and heat-shock protein 90-binding domain; E, activation function 2 transaction domain. |
ER Coactivators and Corepressors
Ligand-bound receptors interact with a family of “coactivator” and “corepressor” proteins that are sensitive to the conformational changes that occur in the LBD of each receptor. These coregulatory proteins interact with the ER to either increase or decrease transcriptional activity at a promoter site. One key mechanism for the coactivator and corepressor modulation of ER transcription likely involves histone acetylation and methylation.26 In addition to alteration of chromatin structure and histone acetylation, coactivators may also promote interactions between the nuclear hormone receptor and the basal transcriptional machinery to activate gene transcription.27 Corepressors have the opposite function and negatively regulate transcription via recruitment of histone deacety-lases.28
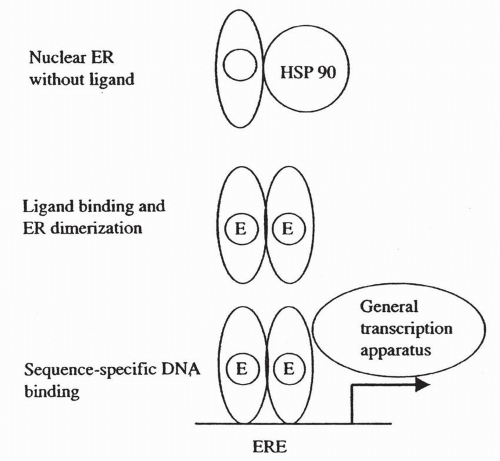 FIGURE 35-5 Simplified operational details of nuclear hormone action, using the estrogen receptor as an example. E, estrogen-occupied ligand-binding site; ER, estrogen receptor; ERE, estrogen response element; HSP, heat-shock protein. |
In general, coactivators and corepressors appear to be expressed at similar levels in many different tissues, suggesting that the responses to estrogen agonists and antagonists are not determined simply by the relative abundance of these cofactors. Instead, it appears that differential regulation of coactivator activity occurs through other signal transduction pathways.26
Nonclassical ER Signal Transduction
ER Activation by Other Signal Transduction Pathways
In addition to classical ER signaling, other ER signaling pathways have been described (Fig. 35-7). Nuclear hormones are not simply receptors for lipid-soluble hormones, but are also critical signaling targets for protein phosphorylation-dependent second messenger pathways.29 These nuclear hormone receptors cross-talk with growth factor pathways that determine the ultimate cellular responses to a complex set of extracellular signals.
 FIGURE 35-6 Consensus core response element motifs for nuclear hormone receptors. AR, androgen receptor; ARE, androgen receptor response element; DR, direct repeat; ER, estrogen receptor; ERE, estrogen response element; GR, glucocorticoid receptor; GRE, glucocorticoid response element; IP, inverted palindrome; n, any nucleotide; P, palindrome; PgR, progesterone receptor; PRE, progesterone receptor response element; RAR, retinoic acid receptor; RARE, retinoic acid receptor response element; T3RE, triiodothyronine receptor response element; VDRE, vitamin D receptor response element. |
ER expression and function are strongly influenced by growth factor signaling (Fig. 35-7B). As a result, ER expression levels correlate with distinct patterns of growth factor receptor overexpression. For example, when ErbB2 or EGFR is activated in experimental systems, ER expression is suppressed. This suggests that EGFR and ErbB2 signaling can bypass the requirement for estrogen for breast cancer cell growth and drive breast cancer cells into an ER-negative, endocrine therapy-resistant state.30
In other circumstances, EGFR and ErbB2 signaling can activate the ER in an estrogen-independent manner. Signaling of these growth factors through the mitogen-activated protein kinase (MAPK) cascade leads to phosphorylation of the Ser118 residue in the ERα AF1 domain.31,32 This, in turn, leads to recruitment of coactivators, allowing ligand-independent gene transcription by the ER.30,33 As discussed in the “Hormonal Resistance” section, insights into the cross talk between the ER and other signal transduction pathways provide a potential strategy for novel therapeutic combinations as well as an approach to new predictive tests for endocrine therapy sensitivity.
Tamoxifen: The First Selective Estrogen Receptor Modulator
High doses of estrogen had long been recognized as effective treatment for estrogen receptor-positive (ER+) breast cancer but serious side effects such as thromboembolism prompted the development of alternative strategies.34 The antiestrogen tamoxifen, a nonsteroidal triphenylethylene synthesized in 1966, was first developed as an oral contraceptive but paradoxically was found to induce ovulation. Activity in metastatic breast cancer was first described in the early 1970s.35 Since then, tamoxifen has become a prototype drug for the hormonal treatment of ER+ breast cancer, with use in prevention and treatment of all stages of the disease. Tamoxifen rapidly became the drug of choice for advanced disease, with response rates ranging from 16% to 56%.36, 37, 38 It became the preferred drug not because it proved better than contemporary alternatives but because it was safe and well tolerated. Tamoxifen’s tolerability was one of the chief reasons for its success in adjuvant and prevention trials, as patients are able to take it for prolonged periods of time with acceptable levels of toxicity.35,39, 40, 41 To date, tamoxifen is the only hormonal agent approved not only for the treatment of invasive breast cancer in the early and advanced settings but also for the prevention of breast cancer and treatment of ductal carcinoma in situ. Widespread use of tamoxifen has likely contributed to the recent decline in breast cancer mortality observed in high-incidence Western countries.42
In addition to its generally antagonist effects on the breast, tamoxifen has estrogen agonist effects on other organs including the endometrium,43, 44, 45 the coagulation system,46,47 bone,48,49 and liver.50,51 Therefore, tamoxifen is correctly described as a selective estrogen receptor modulator (SERM) with organ-specific mixed agonist and antagonist effects. The agonist properties of tamoxifen are illustrated occasionally in advanced breast cancer when “flare reactions,” withdrawal responses, and the experimental demonstration of breast tumor growth stimulated by tamoxifen are evidence that tamoxifen can operate as an agonist in breast tissue under certain circumstances.
Clinical Use
Tamoxifen is used for the treatment of ER+ invasive breast cancer in the neoadjuvant, adjuvant, and metastatic settings. Tamoxifen is also used in the treatment of ductal carcinoma in situ and for breast cancer prevention in high-risk patients.
Tamoxifen Metabolism and Pharmacokinetics
The metabolism of tamoxifen is complex.52 Ten major metabolites have been identified in patient’s sera.53, 54, 55 The most abundant plasma metabolite of tamoxifen is N-desmethyltamoxifen.56 Metabolism of tamoxifen is mediated in the liver by cytochrome P-450-dependent oxidases. The metabolites are excreted largely in the bile as conjugates; tamoxifen dose does need to be reduced in the presence of renal dysfunction.57 There is increasing evidence that endoxifen (4-hydroxy-N-desmethyltamoxifen), a second metabolite of tamoxifen, is most responsible for tamoxifen activity.
The initial plasma half-life of tamoxifen ranges from 4 to 14 hours, with a secondary half-life of approximately 7 days.52,57,60,66
Steady-state concentrations of tamoxifen are achieved after 4 to 16 weeks of treatment.66 While the half-life of endoxifen has not been reported, the biologic half-life of the metabolite N-desmethyltamoxifen is 14 days, with a steady-state concentration reached at 8 weeks. These long half-lives reflect the high level of plasma binding to protein (>99%) and enterohepatic recirculation.66 Only free tamoxifen or metabolites can bind to ERs. Tamoxifen persists in the plasma of patients for at least 6 weeks after discontinuation of treatment.67 Because of the long plasma half-life of tamoxifen, at least 4 weeks are required to reach steady-state levels in plasma. A thorough analysis indicated that a single dose of 20 mg a day is the most appropriate approach to tamoxifen administration.67 Because most tamoxifen is bound to serum proteins, tamoxifen is present in low concentrations in the cerebrospinal fluid,68 suggesting that the response to tamoxifen is likely to be poor in leptomeningeal disease and central nervous system metastasis.
Steady-state concentrations of tamoxifen are achieved after 4 to 16 weeks of treatment.66 While the half-life of endoxifen has not been reported, the biologic half-life of the metabolite N-desmethyltamoxifen is 14 days, with a steady-state concentration reached at 8 weeks. These long half-lives reflect the high level of plasma binding to protein (>99%) and enterohepatic recirculation.66 Only free tamoxifen or metabolites can bind to ERs. Tamoxifen persists in the plasma of patients for at least 6 weeks after discontinuation of treatment.67 Because of the long plasma half-life of tamoxifen, at least 4 weeks are required to reach steady-state levels in plasma. A thorough analysis indicated that a single dose of 20 mg a day is the most appropriate approach to tamoxifen administration.67 Because most tamoxifen is bound to serum proteins, tamoxifen is present in low concentrations in the cerebrospinal fluid,68 suggesting that the response to tamoxifen is likely to be poor in leptomeningeal disease and central nervous system metastasis.
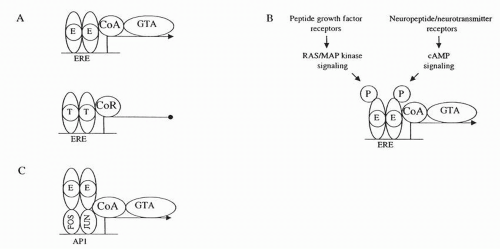 FIGURE 35-7 More complex models of estrogen receptor (ER) models. A. Estrogen (E) promotes coactivator (CoA) interactions that activate the general transcription apparatus (GTA). Tamoxifen (T) promotes corepressor (CoR) interactions that prevent activation of the general transcription apparatus. B. Estrogen receptor is phosphorylated by protein kinases activated by growth factors and neurotransmitters. C. ER interacts with other DNA-binding transcription factors to modulate the transcription of genes that do not possess an ERE. cAMP, cyclic adenosine monophosphate; ERE, estrogen response element; MAP, mitogen-activated protein kinase; P, phosphate. |
Drug Interactions
The CYP3A family is responsible for N-demethylation of tamoxifen (Fig. 35-8). Many other drugs are substrates for this enzyme family, such as erythromycin, nifedipine, cyclosporine, testosterone, diltiazem, and cortisol. The combination of tamoxifen with any of these drugs could potentially interfere with tamoxifen metabolism.69
CYP2D6, which is commonly involved in drug hydroxylation, is the major CYP isoform responsible for the generation of the metabolites 4-hydroxytamoxifen and endoxifen from tamoxifen.70 As discussed in the “Hormonal Resistance” section, a pilot clinical trial of 12 breast cancer patients on adjuvant tamoxifen observed that plasma concentrations of endoxifen appeared to be influenced by the patient’s CYP2D6 genotype.71 The plasma concentrations of endoxifen were significantly lower in patients who were carriers of nonfunctional CYP2D6 allelic variants compared with those who had two functional wild-type alleles.71
The activity of CYP2D6 is also reduced by serotonin selective reuptake inhibitor (SSRI) antidepressants.71,72 In view of the widespread use of SSRIs in patients with a history of breast cancer, a detailed study was done to determine their effects on tamoxifen metabolites.73 Levels of 4-hydroxy-N-desmethyltamoxifen, a metabolite generated via CYP3A4 and CYP2D6 activity, were 64% lower in women taking SSRIs. Other studies also showed that plasma endoxifen concentrations were lower in patients who were also taking the SSRI paroxetine.73 While the clinical significance of these findings is unclear, it is possible that the common combination of tamoxifen and SSRIs may alter clinical results.
Tamoxifen also lowers plasma levels of the aromatase inhibitor letrozole, indicating that at standard doses these two agents should not be administered together.74 Medroxyprogesterone acetate has also been found to alter the metabolism of tamoxifen.75 Reports of an interaction between warfarin and tamoxifen have prompted the manufacturer to list concurrent warfarin use as a contraindication.76 Supratherapeutic effects of warfarin have been reported in patients also receiving tamoxifen; however, the number of cases studied is small. The mechanism of this interaction is likely inhibition of hepatic metabolism of warfarin by tamoxifen. Therefore, close monitoring of coagulation indices is warranted when warfarin and tamoxifen are prescribed together.76
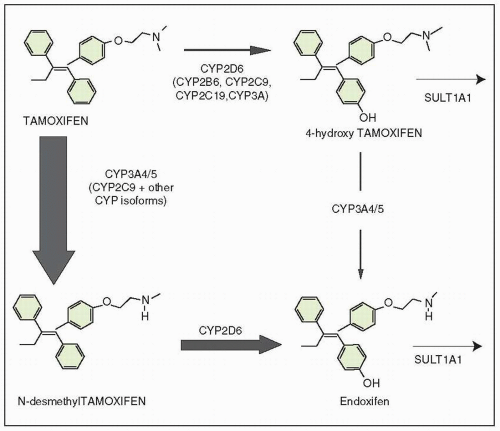 FIGURE 35-8 Selected transformation pathways of tamoxifen and the main CYP enzymes involved. The relative contribution of each pathway to the overall oxidation of tamoxifen is shown by the thickness of the arrow. |
Tamoxifen Side Effects
Although tamoxifen remains a standard hormonal therapy for early-stage and advanced breast cancer, important side effects should be considered. The tamoxifen chemoprevention trial, National Surgical Adjuvant Breast Project (NSABP) P1, established one of the most comprehensive sources of information on tamoxifen toxicity because the true incidence of tamoxifen side effects was not obscured by tumor-related medical problems.77 In NSABP P1, the excess incidence of serious adverse events (pulmonary embolus, deep venous thrombosis, cerebrovascular accident, cataract, and endometrial cancer) for patients receiving tamoxifen therapy was five to six events per 1,000 patient-years of treatment. Other side effects of tamoxifen included hot flashes, nausea, and vaginal discharge. Depression is also commonly considered a side effect of tamoxifen, although there was no clear evidence of this association for women who received tamoxifen or placebo in NSABP P1. In summary, tamoxifen is usually well tolerated and safe and serious side effects occur in no more than 1 in 200 patients annually.
Ophthalmologic Side Effects
Tamoxifen retinopathy, with macular edema and loss of visual acuity, was first reported in patients receiving 120 to 160 mg/d. Three of the four originally reported patients also had corneal opacities.78 The retinal lesions are superficial, white, refractile bodies 3 to 10 μm in diameter in the macula and 30 to 35 μm in diameter in the paramacular tissues, and they occur in the nerve fiber layer, suggesting that they are products of axonal degeneration.79 Additional cases have been described among patients receiving 30 to 180 mg/d.80 Other ophthalmologic findings reported include optic neuritis, macular edema, crystalline macular deposits with reduced visual acuity, intraretinal crystals with noncystoid macular edema, refractile deposits in the paramacular areas with progressive retinal pigment atrophy, bilateral optic disc edema with visual impairment and retinal hemorrhages, tapetoretinal degeneration, and two cases of superior ophthalmic vein thrombosis.80, 81, 82, 83 It is worth emphasizing that, in the chemoprevention study, the only significant optic toxicity at increased incidence compared with placebo was cataract formation.77
Thrombosis
The increased risk of thrombosis associated with tamoxifen therapy may be initiated by decreased levels of antithrombin III levels. Enck and Rios found a decreased functional activity of antithrombin III in 42% of tamoxifen-treated patients.46 Others found reduced antithrombin III and protein C levels in women taking tamoxifen.84 The incidence of venous thrombosis in this study was 5.62%, compared with 0% in controls. In the P1 prevention trial, the average annual rates of stroke, pulmonary embolism, and deep venous thrombosis per 1,000 women were increased by 0.53, 0.46, and 0.50 cases, respectively.77 The NSABP B-14 study reported thromboembolic events in 12 patients (0.9%), with one fatal pulmonary embolus, compared with two thromboemboli (0.2%) in controls,85 a rate similar to that occurring in the P1 prevention trial.77 In the P1 trial, tamoxifen-related thrombotic events were more likely to occur in patients with higher body mass index but not with factor V Leiden or prothrombin gene mutations.86
Hematologic Side Effects
Thrombocytopenia occurs in 5% of patients and is usually transient, resolving after the 1st week of treatment. Leukopenia is less frequent and is also transient.87 The mechanism of these side effects is unclear.
Lipoprotein Effects
Changes in serum lipoproteins in patients taking tamoxifen are indicative of an estrogenic agonist effect: an increase in total triglycerides, a decrease in total cholesterol, an increase in low-density lipoprotein (LDL) triglycerides, and a decrease in LDL cholesterol. Many studies substantiate the effect of tamoxifen on lowering total cholesterol and, in most cases, LDL cholesterol and apolipoprotein B.88, 89, 90 Despite these potentially favorable effects, there was no evidence of an improvement in the rate of cardiovascular mortality in either the Oxford meta-analysis43 of tamoxifen trials or the P1 prevention trial.77 It is possible that beneficial effects of improved lipoprotein levels induced by tamoxifen are counteracted in part by increased incidence of thromboembolism.
Hepatic Toxicity
Tamoxifen has been associated with various hepatic abnormalities, including cholestasis, jaundice, peliosis hepatitis, and hepatitis. Carcinogenicity studies in rats reveal hepatocellular carcinoma in dosages ranging from 5 to 35 mg/kg/d.91 A dose of 20 to 40 mg given to humans is 5- to 10-fold lower than the rat dose range of 5 mg/kg, and an increase in hepatocellular carcinoma in humans taking tamoxifen has not been reported. This is significant because tamoxifen metabolites form adducts with DNA and could be mutagenic and carcinogenic.92
Bone Side Effects
The positive effect of tamoxifen on bone mineral content in postmenopausal women can be considered an advantageous side effect due to its estrogen agonistic activity.93,94 In the P1 study, a nonsignificant decrease in fracture rate was documented.77 In premenopausal women, bone mineral density is decreased, possibly because in a high-estrogen environment, tamoxifen may operate predominantly as an antagonist.93,95
Gynecologic Side Effects
A change in the duration of menses or heaviness of flow, and an increased incidence of ovarian cysts may occur. A small increase in endometrial cancer occurs, but screening for endometrial cancer is complicated by tamoxifen-induced benign endometrial hyperplasia and polyp formation. The risk of endometrial cancer relates to age and duration of tamoxifen treatment. In the original Swedish report, the highest frequency occurred among those treated for 5 years and the Swedish patients received the high dose of 40 mg daily.96 Of 17 tamoxifen-related endometrial cancers, 16 were grade 1 or 2, although three patients died from their endometrial cancer.96 A case-control study found that exposure to tamoxifen interacts with other risk factors for endometrial cancer such as a history of hormone replacement therapy and higher body mass index.97
In addition to endometrial cancer, the risk of uterine sarcoma, a very rare malignancy, is also increased by tamoxifen. Data from six NSABP trials in which over 17,000 women were randomized to tamoxifen or placebo shows that the rate of uterine sarcoma was increased to 0.17 per 1,000 years in women taking tamoxifen versus 0 per 1,000 years in those taking placebo.98
Recommendations for gynecologic follow-up of patients taking tamoxifen have varied from observation to yearly vaginal ultrasound to yearly endometrial biopsy, with no solid data supporting any of these approaches. Yearly pelvic examinations and rapid investigation of postmenopausal bleeding, but not radiologic or biopsy screening, are currently recommended.
Pregnancy
Given the possible teratogenicity of tamoxifen, women should use mechanical contraception while on the drug. Of 50 pregnancies on the file at AstraZeneca Pharmaceuticals in women taking tamoxifen, there were 19 normal births, 8 terminated pregnancies, 13 unknown outcomes, and 10 infants with a fetal and neonatal disorder, 2 of whom had craniofacial defects. Yet in another report, 85 women taking tamoxifen for prevention of breast cancer became pregnant; none of these pregnancies resulted in fetal abnormalities.99 Another case of an infant born with ambiguous genitalia after in utero exposure through 20 weeks has also been reported.100 The evidence for the teratogenicity of tamoxifen is primarily derived from animal studies, in which reproductive organ abnormalities and increased susceptibility to carcinogens were found.101 Nevertheless, tamoxifen is contraindicated in women who are pregnant or planning pregnancy.
Male Breast Cancer
There have been case reports of impotence102 and of nocturnal priapism103 in male breast cancer patients receiving tamoxifen. Another series reported a decrease in libido in 29.2% of male breast cancer patients.104 Impotence has been studied in men treated with tamoxifen for male infertility. One paper reports a loss of libido in four cases (9%),105 whereas studies that reported an increase in testosterone levels with tamoxifen did not report an increase in impotence.106 These data suggest that tamoxifen has minimal effect on sexual function and is probably not the cause of impotence.
Flare Reactions and Withdrawal Responses
Tamoxifen has been characterized as having both estrogen agonist and antagonist actions. These functions are also tissue and perhaps tumor specific. Tamoxifen’s agonist effects on the endometrium, bones, and clotting profile have been described. On the breast its antagonist actions are manifest by anti-breast cancer effects. In patients with advanced breast cancer tamoxifen’s occasional paradoxical effects are manifest by the phenomena of a clinical flare and a drug withdrawal response. A clinical flare reaction is characterized by a dramatic increase in bone pain and an increase in the size and number of metastatic skin nodules with erythema.107 Typically, symptoms occur from 2 days to 3 weeks after starting treatment and can be accompanied by hypercalcemia, which occurs in approximately 5% of patients.108 Tumor regression may occur as the reaction subsides.109
Other SERMs
Background
The structures of other SERMs and antiestrogens are provided in Figure 35.9. Alternative antiestrogens with a modified mixed agonist and antagonist profile have been evaluated with an eye toward developing drugs that are antiestrogenic in the breast and endometrium but retain beneficial effects on bone mineralization. However, although ideal SERMs may represent a small advance in terms of safety, they are not necessarily more efficacious. In general, antiestrogens that
exhibit a mixed agonist and antagonist profile, even if modified in a way that improves tissue-specific toxicities, are likely to exhibit resistance and toxicity profiles that overlap those of tamoxifen.110 This may be because any antiestrogen that triggers ER dimerization and DNA binding is prone to the same coactivator-based resistance mechanisms that may limit the activity of tamoxifen.
exhibit a mixed agonist and antagonist profile, even if modified in a way that improves tissue-specific toxicities, are likely to exhibit resistance and toxicity profiles that overlap those of tamoxifen.110 This may be because any antiestrogen that triggers ER dimerization and DNA binding is prone to the same coactivator-based resistance mechanisms that may limit the activity of tamoxifen.
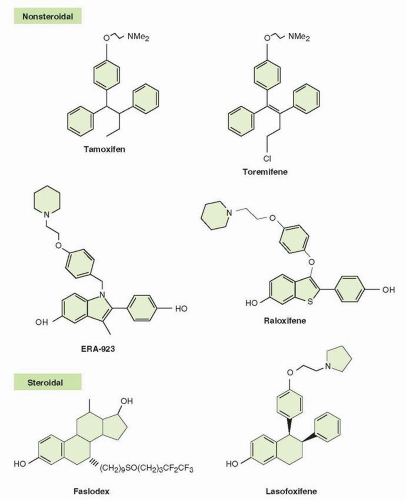 FIGURE 35-9 Antiestrogens. SERM, selective estrogen receptor modulator. |
Raloxifene
Raloxifene [6-hydroxy-2-(4-hydroxyphenyl)-benzo[b]thien-3-yl]-[4-[2-(1-piperidinyl) ethoxy] phenyl] is the first approved drug to exhibit a “modified” SERM profile; however, the first approved indication for raloxifene was osteoporosis, not breast cancer.111 An early evaluation of activity in tamoxifen-resistant breast cancer was disappointing.112 Consequently, raloxifene is not used for the treatment of either early-stage or advanced breast cancer. However, raloxifene has been approved in the breast cancer prevention setting.113 The NSABP-P2 prevention trial demonstrated that in women at high risk for developing breast cancer, raloxifene is as effective as tamoxifen in reducing the risk of invasive breast cancer and has a lower risk of thromboembolic events, endometrial cancer, and cataracts.114 It is important to note that raloxifene has a nonstatistically significant higher risk of noninvasive (in situ) breast cancer compared to tamoxifen, raising the possibility that it may not be as effective in long-term prevention of invasive cancer.
Mechanism of Action
While raloxifene has agonist and antagonist activity similar to tamoxifen in breast tissue and bones, its nil or only very mild agonist effect on the endometrium results in no or very mild increase in endometrial cancer risk, a distinct advantage over tamoxifen. Raloxifene is
a benzothiophene with a structure that includes a flexible “hinge” region. This modification results in a nearly orthogonal orientation of its side chains. This is markedly different from tamoxifen, which has a rigid triphenylethylene structure.115 Although both drugs bind to the ER, their structural differences lead to dissimilar conformations of the ER-ligand complex.116 In the Ishikawa endometrial carcinoma cell line, tamoxifen, but not raloxifene, induces recruitment of coactivators SRC-1, AIB1, and CBP to the c-Myc and IGF-1 promoters.117 This recruitment of coactivators is critical to tamoxifen’s transactivating ability and agonist activity in the endometrium. The comparative inability of raloxifene to assemble this coactivator complex may account for its lower agonist activity in the endometrium.
a benzothiophene with a structure that includes a flexible “hinge” region. This modification results in a nearly orthogonal orientation of its side chains. This is markedly different from tamoxifen, which has a rigid triphenylethylene structure.115 Although both drugs bind to the ER, their structural differences lead to dissimilar conformations of the ER-ligand complex.116 In the Ishikawa endometrial carcinoma cell line, tamoxifen, but not raloxifene, induces recruitment of coactivators SRC-1, AIB1, and CBP to the c-Myc and IGF-1 promoters.117 This recruitment of coactivators is critical to tamoxifen’s transactivating ability and agonist activity in the endometrium. The comparative inability of raloxifene to assemble this coactivator complex may account for its lower agonist activity in the endometrium.
Clinical Use
Raloxifene is indicated for breast cancer prevention in postmenopausal women at high risk for developing breast cancer.
Pharmacology and Metabolism
Raloxifene binds to the ER with a Kd of about 50 pmol/L, which is comparable to the value for estradiol.118 The drug is given at 60 mg/d, and after oral administration it is rapidly absorbed, reaching its maximal concentration in about 30 minutes.119 While absorption is 60%, first-pass metabolism limits the drug’s bioavailability to 2%. After absorption, raloxifene is distributed widely throughout the body and is bound to plasma proteins, including albumin. The half-life of the drug is 27.7 hours.120
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


