Cell of Origin
Molecular studies of isolated tumor cells have demonstrated that lymphocyte-predominant (LP) cells of nodular lymphocyte-predominant Hodgkin’s lymphoma (NLPHL) are derived from antigen-selected germinal center (GC) B cells, whereas Reed-Sternberg (RS) cells in classic HL (cHL) appear to be derived from preapoptotic crippled GC B cells (Table 102.1).6–9 Molecular features of LP cells include the presence of clonally rearranged and somatically mutated immunoglobulin (Ig)V gene cells, with signs of ongoing somatic hypermutation in a fraction of cases (see Table 102.1). These data linked the origin of LP cells in NLPHL to GC B cells. Another important feature supporting this linking was the immunohistochemical expression of BCL6 (a typical GC B-cell marker) in LP cells (Table 102.2).10,11 Accordingly, LP cells can morphologically be observed in an environmental architecture resembling the structure of a secondary follicle, which contains a reactive GC. In fact, in the early phases of NLPHL, LP cells can be found in follicular structures in association with follicular dendritic cells (FDC) and GC type T-helper cells, in this regard resembling GC.12
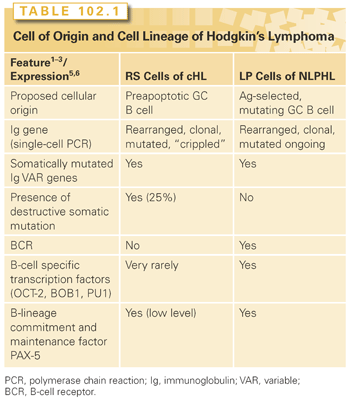

The derivation of NLPHL from GCs is supported by the following features: (1) the expression of the BCL6 gene product and CD40 by LP cells13,14; (2) the occurrence of numerous CD4+/CD57+/PD1 T cells surrounding the LP cells, as seen in normal GCs and progressively transformed GCs (PTGC)6; (3) the presence of an FDC meshwork (CD21+/CD35+) within the tumor nodules15; and (4) the global gene expression profile.16 Conversely, molecular features of RS cells in cHL demonstrate that they are probably derived from GC B cells that have acquired disadvantageous immunoglobulin variable chain gene mutations and normally would have undergone apoptosis. 6–8 In parallel to molecular investigations, biologic markers identifying distinct subsets of mature B cells have been used to study the cell of origin (see Table 102.2). According to the differential expression of these markers, LP and RS cells resemble mature B cells deriving from different stages of B-cell differentiation (i.e., GC and post-GC, respectively).
Reed-Sternberg Cells Lack Common B-Cell Markers
The loss of the B-cell phenotype in RS cells is unique among human lymphomas in the extent to which the lymphoma cells have undergone reprogramming of gene expression. As shown in gene expression profiling (GEP) studies, RS cells have lost the expression of most B-cell–typical genes and acquired expression of multiple genes that are typical for other types of cells in the immune system. Moreover, RS cell gene expression is most similar to that of Epstein-Barr virus (EBV)-transformed B cells, and cell lines derived from diffuse large-cell lymphomas showing features of in vitro activated B cells.17
The deregulated expression of inhibitors of B-cell molecules (inhibitor of differentiation and DNA binding 2 [ID2], activated B-cell factor 1 [ABF1], and notch 1), the downregulation of B-cell transcription factors (OCT2, BOB1, and PU.1), and the epigenetic silencing of B-cell genes (CD19 and immunoglobulin H [IgH]) all seem to be involved in the loss of the B-cell phenotype in RS cells.17,18
Multiple Signaling Pathways and Transcription Factors Have Deregulated Activity in Reed-Sternberg Cells
Very recently, biologic studies on HL cell lines using new technologies have shown that multiple signaling pathways and transcription factors have deregulated activity in RS cells. Involved pathways and transcription factors included nuclear factor kappa B (NF-+B), Janus kinase/signal transducers and activators of transcription (Jak-Stat), phosphoinositide 3-kinase (PI3K)–Akt, extracellular signal-regulated kinase (ERK), activating protein-1 (AP-1), notch 1, and receptor tyrosine kinases.7,19 Functional studies have shown that in normal B GC cells, the activation of the CD40 receptor leads to NF-κB–mediated induction of the interferon regulatory factor 4/multiple myeloma oncogene 1 (IRF4/MUM1) transcription factor. CD40 engagement in HL cell lines by both soluble (s) CD40L and membrane-bound (mb) CD40L upregulates IRF4/MUM1 expression by HL cells.20 CD40 engagement in HL cells by both sCD40L and mbCD40L enhances both clonogenic capacity and colony cell survival of HL cell lines, stimulates proliferation and rescue from apoptosis, mediates in vitro rosetting of activated CD4+ T cells to HL cells, and increases ERK phosphorylation and cell survival.14,21
PATHOLOGY OF HODGKIN’S LYMPHOMA
The REAL Classification and the WHO Proposal
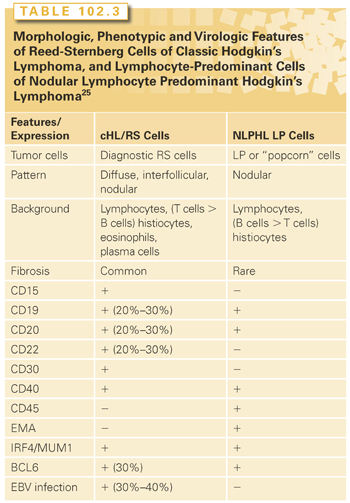
The most recent contribution is provided by the Revised European American Lymphoma (REAL) classification22 and the WHO proposal, which, on the basis of a combination of phenotypic and morphologic features, subdivided HL into two distinct pathologic and biologic entities: NLPHL and cHL. cHL includes four subtypes (see Table 102.2) (see the following). LP cells of NLPHL and RS cells of cHL have different morphology, different phenotype, and different infection pattern with the EBV.22 LP cells express CD20, CD45, and epithelial membrane antigen (EMA) antigens, whereas RS cells display CD15-positive, CD30-positive, and CD45-negative phenotypes. EBV infection is usually present only in the RS cells of cHL, which express EBV encoded latent membrane protein 1 (LMP1) (Table 102.3).22,23
Nodular Lymphocyte-Predominant Hodgkin’s Lymphoma
Morphology
NLPHL is characterized by a nodular, or a nodular and diffuse, proliferation of RS cell variants known as LP cells. LP cells are large and usually have one large multilobated nucleus and scant cytoplasm. The nucleoli are usually multiple, basophilic, and smaller than those seen in classical RS cells.
Phenotype
LP cells are positive for CD20, CD79a, CD75, BCL6, and CD45 and epithelial membrane antigen in nearly all cases (see Table 102.3).11,19 CD75, formerly LN1, is superior to CD20 and CD79a in detecting LP cells. LP cells express CD75 strongly, whereas small reactive B cells in the background show weak cytoplasmic positivity in the Golgi area but no membranous staining, in accordance with their mantle cell phenotype. CD20 is expressed in LP cells equally or less than in the small reactive B cells in the background. CD79a is even worse than CD20 in detecting LP cells because preferentially stains of small reactive B cells. The OCT2, BOB1, PAX5, and PU.1 B-cell transcription factors and the activation-induced deaminase enzyme (which is involved in somatic hypermutation and class switch recombination mechanisms in Ig genes), are consistently coexpressed (see Table 102.1).11,19
Microenvironment
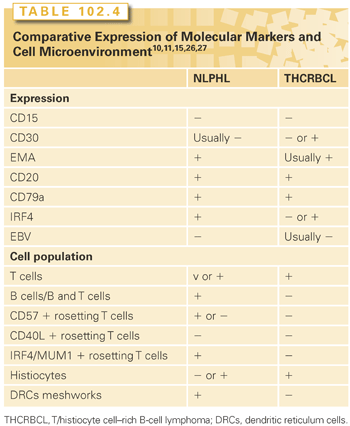
The LP cells reside within nodules consisting of spherical meshworks of FDCs that are filled with nonneoplastic inflammatory cells. Inflammatory cells include small B cells, T cells that specifically express CD3 and CD4, and histiocytes. Furthermore, the inflammatory cells of nodules of NLPHL are characterized by an increase in GC-derived CD57+, IRF4/MUM1+, and PD-1+ T cells (see Table 102.3). Tia1 and CD40L-positive CD3/CD4 positive T cells are absent. PD1 ringing is a feature commonly seen in NLPHL.15,19,24,25
In conclusion, LP cells of NLPHL clearly resemble GC B cells in many phenotypic and genetic aspects, and proliferate in association with a cellular microenvironment that retains key features of a normal GC environment. Although LP cells are found to be even more similar to diffuse large B-cell lymphoma (including T-cell/histiocyte-rich large B-cell lymphoma) in terms of phenotypic and gene expression aspects, the environmental characteristics discriminate between these lymphomas (Table 102.4).
Microenvironment and Histologic Patterns
Well-recognized morphologic features of NLPHL include a nodular, or a nodular and diffuse, proliferation of scattered LP tumor cells, set against a background of reactive lymphocytes reminiscent of a primary follicle. Different patterns are recognizable in NLPHL on morphologic and immunohistologic grounds. Fan and colleagues26 identified six distinct immunoarchitectural patterns (classical nodular, serpiginous/interconnected nodular, nodular with prominent extranodular LP cells, T-cell–rich nodular, diffuse with a T-cell–rich background, and diffuse, B-cell–rich pattern) and two variant patterns (presence of small GCs within the nodules and the presence of prominent sclerosis) (Fig. 102.2). In the nodular pattern originally described by Fan and colleagues26 as pattern A, rare LP cells are seen outside of the nodule. In other patterns, however, increasing numbers of LP cells extend outside of the neoplastic nodules and infiltrate the perinodular space (see Fig. 102.2).26
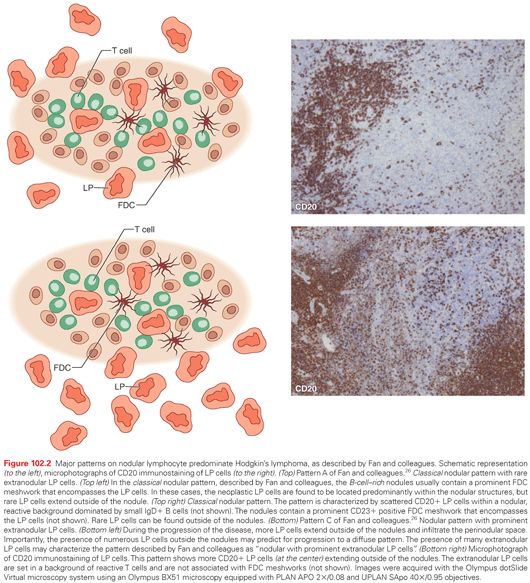
A recent study recognized an additional nodular pattern of NLPHL in which LP cells reside in an environment reminiscent of lymphoid follicles and do not invade the extranodular space (Fig. 102.3).12,27 The recognition of this pattern primarily relies on the identification within the nodules of BCL6+ and CD20+ LP cells, surrounded by rosetting PD1+ T cells. CD23 and CD21 immunostaining usually detects meshworks of FDCs, which entrap the LP cells and the surrounding T-cell rosettes. LP tumor cells are localized within an environment reminiscent of a secondary follicle or, more frequently, within neoplastic nodules reminiscent of a primary follicle without residual GCs (see Fig. 102.3).
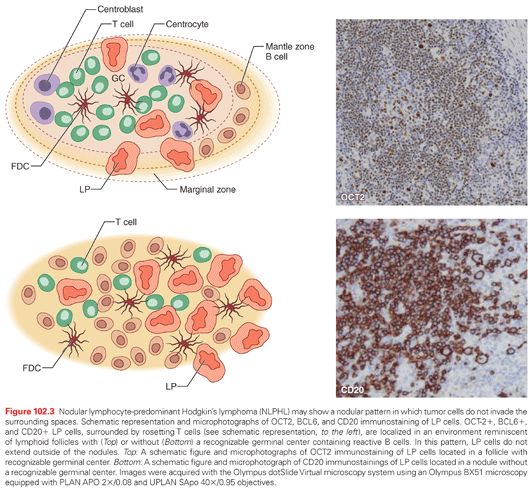
Regarding the relationship of these histopathologic patterns to the clinical course of the disease, the pattern A of Fan and colleagues was usually seen in those patients presenting with earlier clinical stage NLPHL. In general, the clinical impact of the different histopathologic patterns is still uncertain. Understanding this issue is difficult, because more than one pattern is frequently present at the same time.28
Classic Hodgkin’s Lymphoma
Morphology
The so-called RS cell is the diagnostic key for this lymphoma because of its typical morphology: a giant cell with bi- or multinucleation and huge nucleoli. The typical morphology of binucleated and multinucleated RS cells and their mononuclear variant, the so-called Hodgkin’s cell, are not specific to cHL, because they can also be observed in B-NHL (especially in diffuse large B-cell lymphoma [DLBCL] of the anaplastic variant), but they are pathognomonic for cHL in conjunction with an abundant cellular background composed of a varying spectrum of nonneoplastic inflammatory cells.5
Based on the characteristics of the reactive infiltrate, four histologic subtypes have been distinguished: lymphocyte-rich cHL (LRCHL), nodular sclerosis (NS) cHL, mixed cellularity (MC) cHL, and lymphocyte depletion (LD) cHL. LRCHL accounts for only a small fraction (3% to 5%) of all HLs. Most LRCHLs have a better prognosis than do other cHLs and are characterized histologically by a small number of RS cells expressing a cHL immunophenotype. Based on these histologic and clinical features, there is no clear consensus on whether LRCHL represents a distinct disorder or just an early presentation of cHL. On the other hand, LRCHL cases display features intermediate between those of cHL and NLPHL.11,19
Phenotype
Phenotypically, RS cells of cHL are consistently positive for CD30, CD15, CD40, and IRF4/MUM1 (see Table 102.3).23
Microenvironment
cHL is a lymphoid neoplasm, derived from B cells, composed of mononuclear Hodgkin’s cells and multinucleated RS cells residing in an abundant cellular microenvironment. In cHL, microenvironmental cell types include T- and B-reactive lymphocytes, eosinophils, mast cells, histiocytes/macrophages, plasma cells, and granulocytes (Fig. 102.4).25,29–33 In addition, a great number of fibroblast-like cells and interdigitating reticulum cells are detectable, often in association with RS cells, within the collagen bands of NS cHL. Fibrosis—considered a common morphologic feature of HL lesions—is found more frequently in cHL subtypes than in NLPHL. An abnormal network of cytokines and chemokines and/or their receptors in RS cells is involved in the attraction of many of the microenvironmental cells into the lymphoma background (see Fig. 102.4).8,34

Nonmalignant inflammatory/immune cellular components of the HL microenvironment express molecules involved in cancer cell growth and survival, such as CD30L or CD40L, or in immune escape, such as programmed death 1 (PD-1). For example, CD30L+ eosinophils and mast cells, and proliferation-inducing ligand (APRIL)+ neutrophils, are consistently admixed to RS cells, whereas CD40L-expressing CD4+ T lymphocytes rosette RS cells. A considerable fraction of infiltrating CD4+ T cells are regulatory T (Treg) cells. Treg cells and PD-1+ T cells also interact with RS cells, which produce the Treg attractant galectin-1 and the PD-1 ligand (PDL-1).7,25,29 The nonmalignant cells that compose most of the cellular background of cHL are recruited and/or induced to proliferate by tumor cells. They in turn produce soluble or membrane-bound molecules involved in tumor cell growth and survival. Numerous molecules are involved directly or indirectly in the recruitment and/or proliferation of cells constituting the cHL microenvironment. Normal cells may be recruited by cytokines/chemokines produced by RS cells or by T cells and fibroblasts activated by RS cells. RS cells produce molecules capable of inducing proliferation and/or differentiation of eosinophils, Treg cells, and fibroblasts.29
Epstein-Barr Virus Infection
Generally, in the different histologic subtypes of cHL, the immunophenotypic and genetic features of RS cells are identical, whereas their association with EBV shows differences. EBV is found in RS cells in about 40% of cHL cases in the Western world, mostly in cases of MC and LD HL, and less frequently in NS and LRCHL. Conversely, EBV is found in RS cells in nearly all cases of HL occurring in patients infected with HIV.35 Independent studies have recently demonstrated that EBV can transform antigen receptor–deficient GC B cells, which enables their escape from apoptosis. The continued survival of the rescued preapoptotic B cells allows their proliferation. The EBV-encoded latent membrane protein (LMP) 2A is likely to function as the surrogate receptor through which B-cell signaling is triggered. This mechanism of EBV/LMP2A-induced the escape of antigen receptor-deficient GC B cells from apoptosis offers an intriguing model of lymphomagenesis. EBV infection might also affect the microenvironment composition by increasing the production of molecules involved in immune escape and T-cell recruitment, such as interleukin 10 (IL-10), CCL5, CCL20, and CXCL10.36 LMP1 could have an interacting role with the microenvironment. Recent evidence indicates that EBV can manipulate the tumor microenvironment through the secretion of specific viral and cellular components into exosomes, small endocytically derived vesicles that are released from cells.37,38 Exosomes produced by tumor cells from EBV-infected nasopharyngeal carcinoma contain LMP1, which can activate critical signaling pathways in uninfected neighboring cells, suggesting messenger functions of virus-modified exosomes.37 Moreover, in B-cell lines, EBV-modified exosomes would activate cellular signaling mediated through integrins, actin, interferon, and NF-κB.38 Further insights in these mechanisms are emerging from the understanding of the capability of EBV to modulate the (tumor-like) microenvironment.
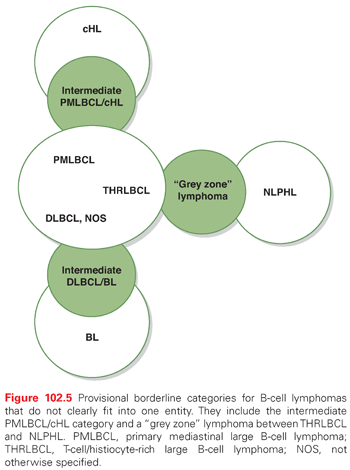
Pathologically, HL subtypes should be distinguished from other B-cell lymphomas showing large and CD30 expressing tumor cells. Figure 102.5 shows B-cell lymphomas, which can be differentiated from NLPHL and cHL on immunophenotypic grounds. The figure also includes lymphomas that have overlapping features with cHL or NLPHL. Most importantly, NLPHL should be differentiated from T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL), a DLBCL subtype, and from the rare cHL variant termed lymphocyte-rich cHL.11,19
Nodular Lymphocyte-Predominant Hodgkin’s Lymphoma
According to current criteria, the detection of one nodule showing the typical features of NLPHL in an otherwise diffuse growth pattern is sufficient to exclude the diagnosis of primary THRLBCL. NLPHL may mimic THRLBCL in a subset of cases in which T cells, rather than B cells, are predominant. This typically occurs in older lesions in which T cells have infiltrated the nodules of B cells and disrupted the nodular architecture.11 This finding was previously termed NLPHL with diffuse areas; the current preferred term is NLPHL, THRLBCL-like. These kinds of lesions have not been associated with aggressive clinical behavior. The presence of small B-cells and CD4+/CD57+ T cells points to a NLPHL diagnosis, whereas the absence of small B-cells, and the presence of CD8+ cells and TIA1+ cells points to primary THRLBCL (see Table 102.4). However, there may be a morphologic and phenotypic gray area between THRLBCL and NLPHL. CD4+/CD57+/PD1 small lymphocytes resetting around typical CD20+/BCL6+ LP cells are useful for the differential diagnosis with PTGC, LRCHL, and THRLBCL. In addition, staining for OCT2, PAX5, and PU1 should be considered as an important diagnostic tool. Interestingly, IgD identifies a subgroup of cases (10% to 20%) with peculiar phenotypical and clinical features.
LRCHL is the most difficult cHL subtype to differentiate from NLPHL, and misclassification has frequently been found in retrospective studies. RS cells in LRCHL can resemble LP cells morphologically; but, immunophenotypically RS cells in LRCHL are positive for CD30 and often express CD15. CD20 can be expressed but is typically weaker and less uniform than CD30 expression. NLPHL is PAX5+, OCT2+, and PU.1+, whereas cHL, including LRCHL, is PAX5+/−, OCT2−, and PU.1−. The distinction between NLPHL and LRCHL is essential, owing to therapeutic and prognostic differences.
cHL
cHL variants should be distinguished from DLBCL subtypes or DLBCL NOS variants that express CD30 (see Fig. 102.5), despite the fact that RS cells have lost much of the B-cell–specific markers. Most or all RS cells also lack the transcription factors OCT2, BOB.1, and PU.1. Instead, RS cells display, in varying frequency, molecules not normally expressed by B cells and B-NHL, such as CD30, CD15, CD70, thymus and activation-regulated chemokine (TARC), A20, fascin, and RANTES.
Finally, cHL cases rich in neoplastic cells may resemble, in particular, large B-cell lymphoma displaying anaplastic morphology and expressing CD30 or primary mediastinal large B-cell lymphoma (PMBCL). There is also a true morphologic and biologic overlap between PMBCL and cHL cases (see Fig. 102.5).
Overlapping Features of PMLBCL with cHL: The So-Called Mediastinal Gray Zone Lymphoma
Mediastinal B-cell lymphomas are mostly represented by NS cHL and PMLBCL. Although PMLBCL and NS cHL have several distinctive pathologic features (Table 102.5),39–42 these entities exhibit strikingly similar clinical presentations (young women with an anterior mediastinal mass) and, in some cases, show overlap in pathologic, genetic, and molecular features (see Table 102.5). A provisional category, designated B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and cHL has been introduced in the WHO proposal to encompass such cases. Table 102.5 shows the main morphologic, phenotypic, and genetic features that may be useful in distinguishing PMLBCL, cHL, and the provisional intermediate category PMLBCL/cHL.

Variant sharing features of DLBCL with anaplastic morphology and cHL may also occur. This shows an expression of CD30, CD15, surface markers, and transcription factors of B cells, commonly absent from RS cells (CD45RB, CD20, CD79, and OCT2).
Molecular Features
In accordance with overlapping phenotypes between cHL and B-cell lymphomas, these lymphomas have a gene expression profile that is intermediate between DLBCL and HL, but closely resembles PMLBCL. Activation of the NF-κB pathway, known to enhance the survival of RS cells, is also a feature of PMLBCL and may represent a survival pathway shared by both neoplasms, likely through the activation of antiapoptotic genes. Activation of the PI3K/AKT pathway were recently identified as a further shared pathogenic mechanism between PMLBCL and cHL. Taken together, molecular features that are common to both lymphoma entities include a decrease of BCR pathway signaling, constitutive NF-κB activation, activation of the cytokine–JAK-STAT pathway, and aberrant activation of the PI3K/AKT pathway. The identification of molecular links between PMLBCL and cHL supports the hypothesis that there may be some pathogenetic overlap between the two entities and that these diseases may in fact represent opposite ends of a continuum.39–42
HIV-Associated Hodgkin’s Lymphoma
The Pre-Highly Active Antiretroviral Therapy Era
In the first years of the AIDS epidemic, HIV-associated HL displayed clinical, pathologic, and biologic peculiarities when compared with HL in people uninfected with HIV. First, HIV-associated HL exhibited unusually aggressive clinical behavior, which mandated the use of specific therapeutic strategies, and it was associated with a poor prognosis. Second, the pathologic spectrum of HIV-associated HL differed markedly from that of HL in people uninfected with HIV. In particular, the aggressive histologic subtypes of cHL, namely MC and LD, predominated among HIV-associated HL.43 Tumor tissue was characterized by an unusually large proportion of RS cells infected by EBV. The fact that LMP1 was expressed in virtually all HIV-associated HL cases suggested that EBV plays an etiologic role in the pathogenesis of HIV-associated HL.
The Highly Active Antiretroviral Therapy Era
People with HIV/AIDS (PWHA) seem to be at increased risk of HL than in first years of the epidemic. HL is presently the most common non–AIDS-defining cancer. Patients infected with HIV, who are modestly immunocompromised due to the improvement in CD4 counts associated with this treatment, are more at risk for the development of the nodular sclerosis subtype.44 In this regard, it has been postulated that with increasing CD4+ T cells resulting from highly active antiretroviral therapy (HAART), the appropriate cellular milieu of cHL, surrounding the RS cells, may again be available. In PWHA with improved immunity, CD4+ T cells provide adequate antiapoptotic pathways and mechanisms for immune escape by tumor cells allowing, in this way, the expansion and maintenance of full expression of the disease, as occurs in cHL among people without AIDS.35,45–47 Alternatively, HL may arise as part of an immune reconstitution syndrome. Hypothetically, RS cell may already be present in severe immunosuppressed patients, and partial restoration may allow for the recruitment of surrounding immune cells and the manifestation of the tumor.48,49 A recent study evaluating the effect of immune reconstitution on HL incidence among a cohort of male veterans infected with HIV ever receiving combination antiretroviral therapy (cART) highlighted that immunosuppression and poor viral control may increase HL risk, specifically during immune reconstitution in the interval post-cART initiation. These findings further suggested an immune reconstitution–type mechanism in HIV-related HL development.50
EARLY-STAGE HODGKIN’S LYMPHOMA
The management of early-stage Hodgkin’s lymphoma exemplifies several important principles of oncology. These include the progressive improvement of cure rates through careful clinical research; the identification of prognostic features and new markers of optimal response; the refinement of treatment by the exploration of multimodality approaches; the vital importance of long-term follow-up; and a holistic analysis of the outcomes of treatment. Overall, this is one of the success stories of modern oncology, with modern treatment achieving high initial cure rates (up to 90% with the first-line of therapy) and good overall survival at around 95% after 5 years or more. Because it most often affects younger people in the 2nd to 4th decade of life, this has important implications for the goals of treatment, which must include not only the maximization of initial tumor control but also the avoidance of preventable long-term side effects.
Prognostic Features
The relatively orderly progression of cHL has long been recognized.51 It generally develops through involvement of adjacent nodes in the same anatomical site, then in adjacent nodal areas, and it is extremely rare to find isolated deposits in two distant nodes. The same is not true for nodular lymphocyte-predominant disease, which, in this respect, more closely resembles a low-grade non-Hodgkin’s lymphoma: It often presents with a single isolated node in the neck, but if it does progress, the dissemination is often to distant sites without intervening nodal involvement.
The predictable spread of cHL has allowed for the construction of a staging system based on anatomical extent, so that early-stage disease is defined by involvement of nodal groups on one side of the diaphragm only, more usually the thorax. Stage I disease is confined to a single anatomical nodal group (cervical, supraclavicular, axillary, anterior mediastinal, etc.), whereas a disease affecting more than one such group is stage II.
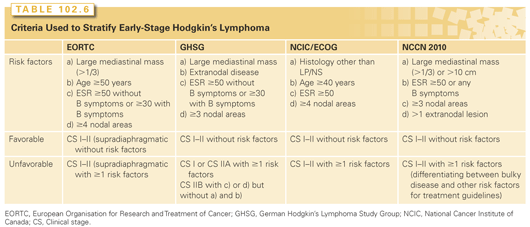
Beyond this division on the basis of nodal involvement, many studies have identified further prognostic features through retrospective analyses of large series of patients in clinical trials, mostly treated with extended field radiotherapy. This has allowed for the subdivision of early-stage disease into favorable and unfavorable categories. These do not represent biologically distinct processes, but act as a useful indicator of the severity of the illness and its optimum management, even though the current approaches to treatment are different to those in use when the factors were identified. Although a variety of stratification systems have been devised, common features include the presence of bulky disease (usually in the mediastinum), more advanced age (with a cutoff of 40 or 50 years of age), elevated erythrocyte sedimentation rate (ESR), systemic symptoms, and multiple or extranodal sites of involvement (Table 102.6).
Radiation Therapy
The effective treatment of HL by radiotherapy began with the work of Gilbert in the 1920s.52 He introduced the rationale for treating both the evident sites of nodal involvement and adjacent but clinically uninvolved lymph nodes, on the basis that these were likely to contain microscopic disease. Peters53 took the same approach at the Princess Margaret Hospital in the 1940s, publishing a landmark paper in the American Journal of Roentgenology in 1950, which described the cure of limited HL by high-dose, fractionated radiation. She reported 5- and 10-year survival rates of 88% and 79%, respectively, for patients with stage I disease, which transformed the outlook for an illness previously thought to have no long-term survivors.
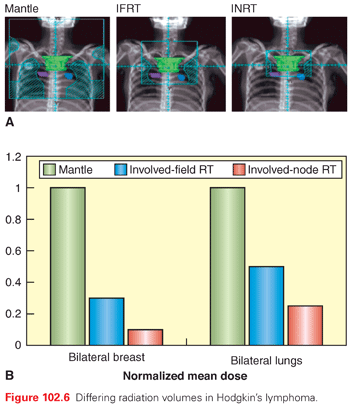
In the early days, radiation therapy utilized fields that included the entire lymphatic system, total lymphoid irradiation (TLI), to relatively higher biologic radiation dosages compared to contemporary treatment. Extended field radiation therapy (EFRT) included all nodal sites using three radiation fields classically known as mantle, para-aortic–spleen, and inverted Y. A variation of EFRT was also used known as subtotal nodal irradiation (STNI).54 This was effective, and in many cases curative, but was accompanied by important long-term toxicities, especially the induction of second malignancies and accelerated cardiovascular disease.55–60 It remained the principal approach to treatment of early disease until clinical trials demonstrated that a combination strategy with chemotherapy could produce superior cure rates with much less irradiation, leading to a reduction of the irradiated field size to only the involved field (IF); the latter was based on a series of studies aimed at minimizing the toxicity of radiation therapy treatment. The German Hodgkin’s Lymphoma Study Group (GHSG) HD8 showed in a randomized trial that reducing the treatment volume from EFRT to involved field radiation therapy (IFRT), when combined with chemotherapy, is equally effective. The European Organisation for Research and Treatment of Cancer (EORTC) H7 showed a similar outcome comparing IFRT to STNI.61,62
The developments in functional imaging, treatment planning, and image-guided radiation therapy have made it possible to better define and further decrease the radiation fields. Thus, IFRT, which is based on anatomic landmarks and encompassing adjacent uninvolved nodal stations, is no longer appropriate. Based on the fact that most recurrences occur in the original nodal sites, involved node irradiation therapy (INRT) was suggested; the field, in this case, is confined to the macroscopically involved nodes on imaging studies at diagnosis. Although this requires a significant margin around the node to allow and ensure adequate coverage, it can still result in significantly lower exposure to adjacent critical structures.63 No formal comparison has been made to the results with IFRT, but multiple studies have shown no loss of efficacy with INRT (Fig. 102.6).64,65
Using INRT requires acquiring images at diagnosis in treatment positions and prior to the start of chemotherapy to minimize anatomic position variations between diagnostic and radiation treatment planning imaging. Because that is not practical in most cases, new guidelines defining involved site radiation therapy (ISRT) has been introduced by the International Lymphoma Radiation Oncology Group (ILROG). The new standard of care represents a significant reduction in the volume included in the previously used IFRT by using modern imaging and radiation planning techniques to limit the amount of normal tissue being irradiated.
Combined Modality Therapy
The recognition that HL is highly sensitive to cytotoxic chemotherapy led to the testing of systemic treatment in early stage disease. By administering limited doses of chemotherapy, it has been shown possible to reduce both the extent and dose of radiotherapy, while still maintaining high cure rates.66–68 The success of this approach has depended on the different treatment of favorable and unfavorable disease, with results in favorable groups excellent even after low impact chemotherapy, such as two cycles of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) or the attenuated EBVP regimen. The EORTC H7-F study compared STNI to six cycles of EBVP followed by IFRT (36 to 40 Gy), with better results from the combined modality treatment: 10-year event-free survival was 88% versus 78%, and overall survival was 92% in both arms.62 The GHSG HD10 study in favorable early disease compared results in a 2 × 2 randomization between two or four cycles of ABVD and 20 or 30 Gy of IFRT. All four groups had very high cure rates, with progression-free survival of 92% and overall survival of 97% at 5 years,69 suggesting that two cycles of ABVD and 20 Gy of IFRT is sufficient treatment for carefully selected favorable disease.
A slightly different picture has emerged from studies of unfavorable early disease, where many patients present with bulky mediastinal nodes. Here, there is a threshold of treatment intensity below which the results become less favorable, with an apparent interaction between the efficacy of chemotherapy and the dose of irradiation used. Attenuated use of either modality can be compensated by the other, but if both elements are reduced too far, the freedom from treatment failure is lowered as the result of the excess of early recurrences. The EORTC H8-U trial showed the equivalence of either six or four cycles of MOPP-ABV when given before IFRT (36 to 40 Gy), or four cycles of MOPP-ABV before STNI, with 5-year event-free survivals of 84%, 88%, and 87%, respectively, and 10-year overall survival estimates of 88%, 85%, and 84%, respectively, indicating that treatment more intensive than four cycles of MOPP-ABV and IFRT was unnecessary, and that less toxic treatment might be possible.70 More recently, the GHSG HD11 study has tested a 2 × 2 randomization between four cycles of ABVD and four cycles of the baseline bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone (BEACOPP) regimen before either 20 Gy or 30 Gy IFRT. The least intensive arm, four cycles of ABVD and 20 Gy, showed inferior 5-year progression-free survival at 82%, compared to 87%, although overall survival was unaffected, at 94.5%.71 This suggests that for the unfavorable early-stage group, it may hazardous to reduce treatment below a threshold of four cycles of ABVD and 30 Gy IFRT, unless some means can be found to select those patients for whom further deintensification can be attempted, such as the use of functional imaging.
Chemotherapy Alone
Recognition of the long-term toxicity of extended field irradiation has led many investigators to test approaches by which radiotherapy may be omitted altogether from the treatment of early HL.72,73 Two large randomized trials have been performed, in pediatric and adult patients, respectively, and both demonstrated that the omission of radiotherapy slightly reduced control of the disease, reflected in lower progression-free survival, but had no adverse impact on overall survival.
The North American Children’s Oncology Group study CCG 5942, tested the omission of low-dose IFRT (21 Gy) for those in complete remission after four cycles of COPP-ABV chemotherapy. The study was closed prematurely when an interim analysis showed a difference in the progression rates in the two arms. With a median 7.7 years follow-up, the event-free survival favored the radiotherapy group (93% versus 83%; p = 0.004), with most recurrences in the chemotherapy-alone group seen at the sites of original disease. There was, however, no difference in overall survival, estimated at 97% at 10 years74.
In adults with early-stage nonbulky disease, the intergroup Eastern Oncology Cooperative Group (ECOG)/National Cancer Institute of Canada (NCIC) study tested treatment with ABVD alone to either 35 Gy STNI in favorable disease, or two cycles of ABVD followed by STNI in unfavorable cases. The first report of this study, with a median follow-up of 4.2 years, showed inferior freedom from progression in the chemotherapy-alone arms (87% versus 93%), with the unfavorable group particularly disadvantaged by the omission of radiotherapy.75 The initial analysis showed no difference in overall survival, but with longer follow-up, a different picture emerged, with inferior 10-year survival among the patients who had received radiotherapy (87% versus 94%, respectively; p = 0.04). The risk of death from lymphoma was not different between the arms, but the risk of death from other causes was more than threefold higher among those treated with radiotherapy, and much of the excess was due to second cancers.76 It is important to note, however, that this protocol involved much more extensive irradiation than is currently in use, making extrapolation of the results difficult.
In the absence of direct comparative trials between modern combined modality therapy and chemotherapy alone, a meta-analysis was performed using the intergroup study ABVD-alone group and the comparable patients from the GHSG HD10 and HD 11 studies who received ABVD and IFRT. This showed that the short-term disease control was inferior with ABVD alone, reflected in worse 8-year time to progression (93% versus 87%; hazard ratio [HR], 0.44; 95% confidence interval [CI], 0.24 to 0.78), but that overall survival was not adversely affected in these groups, with 95% alive in the long-term follow-up.77 The impact of combined modality treatment was particularly apparent among patients who showed less than complete remission after chemotherapy, suggesting that some means of selecting those with chemosensitive disease for deescalation of therapy would be attractive, and might allow radiotherapy to be omitted without a loss of disease control.
Response-Adapted Treatment
Much interest has been generated in the possible use of functional imaging to give an early indication of chemosensitivity in HL. The technique most widely tested is 2-(18F)fluoro-2-deoxy-D-glucose positron emission tomography (FDG-PET), the application of which as an interim readout of efficacy has been enhanced by the development of a highly reproducible five-point scale for reporting the results (Table 102.7).78 This approach appears to improve the sensitivity for the detection of residual active lymphoma when compared to conventional computed tomography,79 but the data from prospective randomized studies using it as a guide to therapy are not yet mature enough for firm conclusions, and it is clear that there is a small but definite false-negative rate for FDG-PET, probably of the order of 5% to 10%.

Two studies have reported early results, with broadly similar outcomes (Table 102.8). The United Kingdom National Cancer Research Institute RAPID study randomized patients with nonbulky early-stage disease who had an interim PET score of 1 or 2 after three cycles of ABVD to either 30 Gy IFRT or no further therapy, and found that the 3-year progression-free survival and overall survival were not significantly different.80 There was, however, a trend toward inferior disease control, which became significant when patients who did not receive the radiotherapy as allocated were excluded (97% versus 90.7%; HR, 2.39; p = 0.003). Similarly, the EORTC H10 study compared two strategies of therapy: standard treatment with ABVD and IFRT, stratified according to baseline prognostic factors, versus a nonradiotherapy approach, but using further chemotherapy, for those with negative FDG-PET scans after two cycles of ABVD.81 The results with a short follow-up suggested inferior disease control in the experimental PET-directed arms, although the number of progressions was small and a much longer follow-up will be required to determine whether there is any detrimental effect on survival.
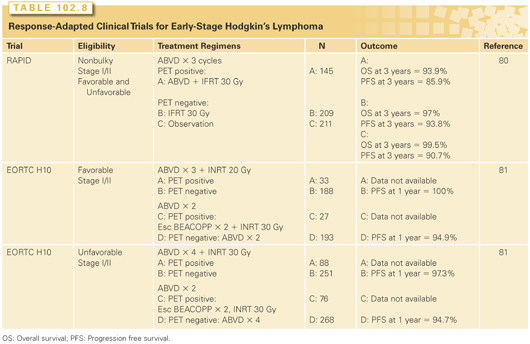
Taken overall, the evidence suggests that for early HL, the use of combined modality treatment produces optimum results in terms of disease control, with a very high expectation of cure from the initial therapy. There is, however, a large proportion of patients (around 90%) who will be curable with chemotherapy alone, and the number needed to treat with radiation in order to achieve 1 extra cured patient is between 15 and 30 according to these trials. Given these figures and the perceived risks of late toxicity from radiotherapy, many patients may prefer the slightly higher risk of recurrent lymphoma to the potential for longer term morbidity. This will, of course, be subject to other variables such as their age, the sites of involvement (and thus the radiotherapy fields), and their baseline risk category. In general, the results of treatment from either approach are very good, and it is reassuring that in almost all the trials carried out, a small reduction in disease control does not have any detrimental effect on overall survival, thanks to the excellent results of second-line therapy, when it is required.
ADVANCED-STAGE HODGKIN’S LYMPHOMA
In patients with advanced-stage HL (stages IIB to IV), the introduction of more effective and less toxic front-line treatment regimens during the last few decades has steadily improved the prognosis. However, complete remissions after initial therapy are not achieved in approximately 20% of patients with stage III to IV disease, eventually leading to disease progression. The current clinical challenge in patients with advanced stage disease is to increase the number of patients with durable remissions and a favorable outcome after initial treatment, while decreasing the incidence of long-term toxicities. The identification of poor prognostic features may allow for a risk-adapted approach to therapy to potentially increase the likelihood of cure and also to minimize side effects.
In general, ABVD chemotherapy remains the most widely used treatment for newly diagnosed patients with advanced-stage HL in the United States. Dose-intense regimens such as escalated BEACOPP are more commonly used in Europe, but are also considered in North America in patients with multiple poor prognostic factors. The future management of advanced-stage HL patients, however, is being shaped by PET-directed approaches and the incorporation of novel agents into these standard combinations.
Prognostic Factors in Advanced Disease
The presence of adverse prognostic factors at diagnosis is one of the methods used to select therapy in HL and the International Prognostic Score (IPS) is an established risk stratification system for advanced disease patients (Table 102.9).82 This prognostic model was constructed using seven factors associated with a poor outcome (serum albumin less than 4 g per deciliter, hemoglobin less than 10.5 g per deciliter, male sex, age 45 years or older, stage IV disease, leukocytosis of at least 15,000 per cubic millimeter, and lymphocytopenia of less than 600 per cubic millimeter or less than 8% of the white cell count). Although the IPS is highly predictive of freedom from disease progression, it does have limitations. The IPS does not adequately define the truly high-risk patients, because only 7% of the patients in the original study were in the high-risk group and their failure-free survival (FFS) at 5 years was still quite reasonable at 42%.82 Furthermore, treatment strategies and supportive care have changed since the development of this prognostic model, and although the IPS is still clearly predictive of outcome, its performance may not be as good as originally described.83–85

Therefore, efforts have been made to improve the prognostication of the IPS by the incorporation of additional clinical prognostic factors,86,87 the inclusion of biologic parameters,88–99 or the addition of an early disease response assessment.100,101 Biologic factors that have been studied include molecular profiling of the tumor and the RS cells88,93–95; the measurement of circulating cytokines or receptors including IL-10, CCL17, or soluble CD3089–92,98; and the enumeration of immune cells such as macrophages in the tumor microenvironment.96,97,99 Although many of the biologic factors have prognostic significance independent of the IPS, they have not been adopted in everyday practice due to issues of reproducibility and a lack of prospective validation. On the other hand, an early response assessment as measured by an interim PET/computed tomography (PET/CT) scan has been shown to be a very powerful prognostic tool that is independent of clinical and biologic prognostic factors, including the IPS.100–102 Interim PET/CT scanning has been introduced into standard clinical practice and is also being utilized as a method to direct treatment choices.
Choice of Initial Therapy
Combination chemotherapy forms the basis of treatment for patients with advanced-stage HL (Table 102.10). Initially, the MOPP regimen (nitrogen mustard, vincristine, procarbazine, and prednisone) was developed for previously untreated patients with very advanced HL, and a long-term follow-up of patients treated with the MOPP regime has confirmed that this combination can cure advanced HL. MOPP resulted in a freedom from progression rate of 54% and an overall survival of 48 % at 20 and now 40 years.103 Although the MOPP regimen had a significant impact on the survival of patients who may previously have died of progressive disease, at least one-third of patients relapsed after MOPP chemotherapy and long-term complications were frequently seen in patients who received the combination.
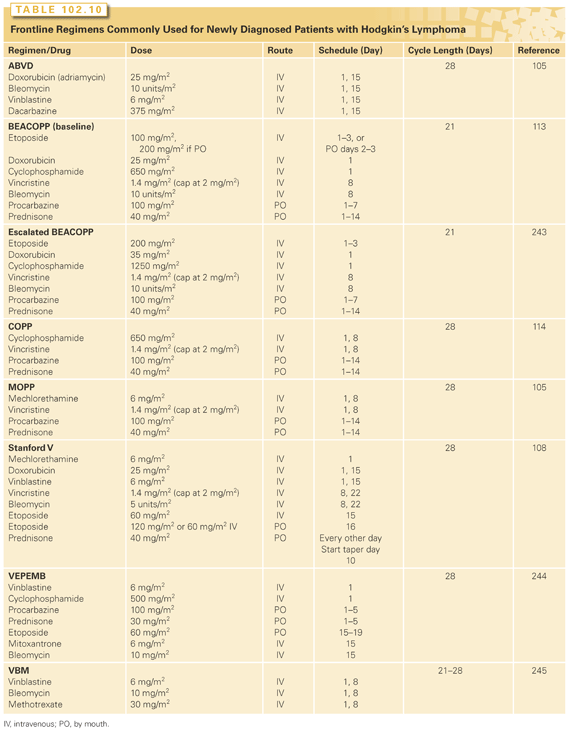
To improve patient outcomes and decrease toxicity, other chemotherapy combinations such as ABVD were developed. An initial randomized trial compared alternating cycles of ABVD and MOPP chemotherapy to a dose and scheduled modified MOPP chemotherapy, and the alternating regimen was found to be superior in respect to the complete remission rate, freedom from progression, and overall survival.104 Subsequently, a number of randomized trials were performed using ABVD in combination with MOPP chemotherapy or using ABVD alone. MOPP, ABVD, and MOPP alternating with ABVD were compared and the complete response rate and freedom from progression was initially found to be superior in patients receiving ABVD or the alternating program, but subsequent follow-up reports of the study show no difference in disease-free or overall survival when compared to a MOPP program also given at reduced doses.105 Two further studies compared the MOPP/ABVD hybrid regimen to MOPP alternating with ABVD, and the regimens were found to be equivalent.106,107 When the MOPP/ABV hybrid regimen was compared to ABVD, ABVD chemotherapy was found to be superior with less toxicity.83 The results of these trials led to ABVD chemotherapy being regarded as a standard of care for patients with advanced HL based on the clinical efficacy of the combination, the ease of administration, and the acceptable toxicity profile.
As an alternative to ABVD, the Stanford V regimen was developed as a short duration regimen combined with radiation therapy.108 The initial single institution results with the regimen showed excellent results with a 5-year freedom from progression of 89% and an overall survival of 96%. These promising results were confirmed in a multi-institutional study.109 The Stanford V regimen has subsequently been compared to ABVD in a number of randomized trials. Initial studies suggested that ABVD might be superior to Stanford V with a 10-year failure-free survival that was superior in ABVD treated patients; however, it has been argued that the differences in outcome may be due to the fact that radiotherapy in the Stanford V arm was administered differently from what was originally described.110 Two subsequent randomized trials comparing ABVD to Stanford V have found no difference in response rate, failure-free survival, or overall survival between the regimens.111,112 Overall, ABVD is felt to be superior to Stanford V in patients with advanced disease.
The GHSG also developed new regimens for patients with advanced HL, particularly standard dose and dose-escalated BEACOPP.113 A randomized trial comparing COPP (cyclophosphamide, vincristine, procarbazine, and prednisone) alternating with ABVD to escalated or standard BEACOPP showed that patients receiving escalated BEACOPP had improved disease control and overall survival.114 The improvement in outcome for patients treated with escalated BEACOPP was sustained with long-term follow-up.115 Although these results were encouraging, long-term complications including acute myeloid leukemia or myelodysplastic syndrome appeared to be more frequent in patients treated with escalated BEACOPP. A similar Italian study compared six cycles of ABVD to four cycles of escalated BEACOPP followed by two cycles of standard BEACOPP and to six cycles of a multidrug intensive regimen. When the results from the ABVD arm were compared the BEACOPP arm, there was an improved progression-free survival with BEACOPP, but the overall survival was not different. Although more toxicity was seen in the BEACOPP-treated patients, poor-risk patients tended to benefit most when treated with BEACOPP.116
Since these initial studies, a number of randomized studies have been performed to determine the optimal number of cycles of BEACOPP needed to maintain the clinical benefit but potentially decrease toxicity, and also to define the subgroup of patients most likely to benefit from a more intensive treatment approach. In a study restricted to those younger than 60 years of age, the GHSG found that six cycles of escalated BEACOPP followed by radiotherapy to PET-positive masses was more effective in terms of freedom from treatment failure and less toxic than eight cycles of the same regimen.117 This led the GHSG to conclude that six cycles of escalated BEACOPP is their standard for advanced HL. To determine whether the high-risk group of patients are those who benefit most from escalated BEACOPP, the EORTC 20012 trial randomized advanced-stage Hodgkin patients with an IPS ≥3 to either eight cycles of ABVD or four cycles of escalated BEACOPP followed by four cycles of standard or baseline BEACOPP.118 At a median follow-up of 3.9 years, event-free survival, which was the primary endpoint, was similar between treatment arms. Although more relapses were observed with ABVD treatment, early discontinuations were more common in BEACOPP-treated patients. In this high-risk group of patients, however, overall survival was not significantly improved with the use of BEACOPP.
Treating physicians who favor using escalated BEACOPP as initial therapy for advanced-stage HL have pointed to the high response rate and improved event-free survival as the reason to use this combination. In contrast, those who favor using ABVD as initial therapy have cited the complication rate with escalated BEACOPP, as well as the ability to salvage relapsing patients with stem cell transplantation, as reasons to use a less intensive treatment first. To compare these approaches, a randomized comparison of ABVD and escalated BEACOPP was reported, but the analysis included second-line therapy if administered.119 Patients with residual or progressive disease after initial ABVD or escalated BEACOPP were treated with salvage therapy, including stem cell transplantation. The authors analyzed the outcome after initial therapy, but also analyzed the outcome after salvage therapy. The freedom from first progression significantly favored patients receiving escalated BEACOPP when compared to patients treated with ABVD (85% compared to 73%; p = 0.004). However, after completion of all planned therapy including salvage therapy for those with residual or progressive disease, the 7-year rate of freedom from second progression was not significantly different (88% in the escalated BEACOPP group and 82% in the ABVD group; p = 0.12) and the 7-year overall survival rate was 89% and 84%, respectively (p = 0.39). Severe adverse events were more commonly seen in patients receiving escalated BEACOPP. These results have led some to suggest that initial therapy may not need to be highly aggressive in all patients due to the fact that relapsing patients may be salvaged with subsequent intensive therapy.120 Others have pointed out that overall survival was a secondary endpoint in this study and that the study was small compared to other similar trials.121 In an attempt to clarify whether a survival difference exists, a meta-analysis was performed that suggested that six cycles of escalated BEACOPP may well improve overall survival when compared to ABVD.122
Overall, it is clear that escalated BEACOPP has greater efficacy than ABVD in patients up to 60 years of age, although escalated BEACOPP-treated patients experience more toxicity, particularly if they are in the upper segment of this age range. Acute and long-term toxicity may, however, be improved by the use of six rather than eight cycles of escalated BEACOPP. It is also clear that approximately two-thirds of patients with advanced HL may not need intensive therapy such as escalated BEACOPP, because they will be cured with ABVD. Clinical risk factors, treatment burden and cost, fertility issues, the risk of long-term relapses, as well as potential short- and long-term complications should be considered as physicians and patients decide which regimen to use as initial treatment for advanced-stage HL.
Positron-Emission Tomography–Directed Approaches
A strategy to potentially optimize therapy for HL, by possibly increasing efficacy and decreasing toxicity, is to utilize PET scans during treatment. Because changes in glucose metabolism precede changes in tumor size, responses can be assessed earlier during treatment with PET scans than with CT. Early interim PET scan imaging after chemotherapy for HL has been shown to be a sensitive prognostic indicator of outcome in patients with advanced disease.123 In prospective studies, interim PET scans after two cycles of ABVD chemotherapy was a significant predictor of progression-free or event-free survival in patients with advanced-stage disease.101,102 Similar findings were reported for patients treated with Stanford V or escalated BEACOPP.124,125 Current clinical trials are now testing whether patient outcomes can be improved by modifying treatment based on the interim PET scan results. In patients who have an inadequate response based on the interim PET scan, treatment is either intensified or salvage therapy is contemplated.
Initial studies testing whether deescalation to less intense or abbreviated therapy maintains efficacy in patients who have a complete response by the interim PET scan suggest that this approach is feasible. Avigdor et al.126 treated advanced-stage HL patients with two cycles of escalated BEACOPP and deescalated to ABVD chemotherapy for four cycles if the PET scan after the initial two cycles was negative. Patients who did not achieve a negative scan were removed from the study and considered for salvage therapy followed by high-dose chemotherapy and autologous stem-cell transplantation. Seventy-two percent of patients had a negative scan, and deescalation to ABVD resulted in a 4-year progression-free survival of 87%.126 In a similar fashion, the GHSG HD18 trial is testing whether the number of cycles of escalated BEACOPP can be reduced from six to four in patients with a negative interim PET scan.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


