Hodgkin lymphoma (HL) is a lymphoproliferative malignancy that accounts for only 1% of newly diagnosed malignancies in the United States; however, the disease’s importance to the field of medical oncology is out of proportion to its clinical incidence. From an historical point of view, HL was the first cancer in which the curative potential of combination chemotherapy was demonstrated. Second, because affected patients are often young, there is a great potential for adding years of productive life by giving curative therapy. Third, because patients with HL are often cured, HL serves as a clinical laboratory for investigating the late effects of cancer therapy.
HL usually presents as solitary or generalized lymphadenopathy and most commonly occurs in young adults, although persons in any age group may be affected. The disease tends to spread in a contiguous fashion, and most patients present with disease limited to the lymph nodes or to the lymph nodes and spleen. Even patients with advanced disease have a reasonable chance of cure. Overall, cure can be achieved in approximately 80% of patients with HL. Treatment of limited disease often incorporates radiation therapy and combination chemotherapy, whereas treatment of advanced disease is generally limited to combination chemotherapy alone.
The evaluation of data and conclusions from clinical trials in HL requires certain caveats. The first issue is that salvage therapy for relapsed HL patients has a relatively high rate of cure. This fortunate fact can complicate the interpretation of trials of initial therapy for HL. In contrast to the situation in many other areas of oncology where salvage therapies generally do not lead to cures, progression-free survival (PFS) in HL trials must not be used as a surrogate marker for overall survival (OS). Conversely, because late complications, including malignancy, can compromise the long-term efficacy of treatments that appear superior in the short run, therapies that produce superior PFS at 5 years may not be associated with superior OS at 15 years. In some situations, then, treatment plans with built-in contingencies may be appropriate. As an example, consider two hypothetical treatment strategies: treatment A is moderately effective but nontoxic, and failures can be salvaged effectively with treatment X. The alternate treatment, B, is very effective but failures are only rarely salvaged with treatment Y. The combination of the (less-effective) treatment A and salvage with × if needed may lead to superior long-term results as opposed to starting out with the (more effective) treatment B and salvage with treatment Y. This may be especially true if one considers short- and long-term toxicities of treatment. When considering initial therapy for patients with HL, one must realize that valid alternatives exist in addition to what may be considered the “best therapy” based on short-term observations, and that individualized approaches may be appropriate to accommodate individual risks, especially for long-term toxicity.1
EARLY HISTORY
In 1832, Thomas Hodgkin presented a paper entitled, “On Some Morbid Appearances of the Absorbent Glands and Spleen.”2 His report was an autopsy description of seven patients, and the major original thesis presented in the paper was that the entity he was describing was a primary process involving the lymph glands and spleen rather than a reactive inflammatory condition. In 1856, Samuel Wilks published a series of cases involving enlargement of the lymph glands3 and noted Hodgkin’s original description. In 1865, Wilks wrote, “Cases of Enlargement of the Lymphatic Glands and Spleen (or Hodgkin’s Disease) with Remarks,” updating and extending his findings.4 Thus, Thomas Hodgkin’s name became linked to the disorder. Of note, some of the cases included in Hodgkin’s initial report were cases of tuberculous lymphadenopathy and non-Hodgkin lymphoma.
After these gross pathologic descriptions, the first microscopic description of HL was reported by Langhans5 in 1872. This report was followed by independent reports by Sternberg in 18986 and by Reed in 19027 describing the characteristic giant cells that came to be known as (RS) cells. At the time of these early reports, all comments regarding the cause of HL were purely speculative. Not surprisingly, these early authors were divided over whether HL represented an infectious disease, an inflammatory disorder, or a malignancy involving the lymph glands.
EPIDEMIOLOGY AND ETIOLOGY
It is estimated that 9,060 new cases of HL will be diagnosed in 2012.8 The male-to-female ratio is 1.2:1.8 In most economically developed countries, there is a bimodal age distribution, with one peak occurring in the third decade of life and the second, smaller, peak occurring after age 50 years.9 The occurrence of HL in patients between the ages of 15 and 39 has been positively associated with increased maternal education, decreased numbers of siblings and playmates, and single-family dwellings in childhood.10 In less economically developed countries, HL is less common but affects children, most of whom are boys; mixed cellularity HL and lymphocyte-depleted HL are more commonly seen.11 These data have been interpreted as supporting the hypothesis that HL is caused by an infectious agent, and it has been postulated that malignancy is more likely to occur when exposure to the agent in question is delayed until late adolescence or early adulthood.11, 12, 13, 14
Epstein-Barr virus (EBV) has been proposed as contributing to the development of some cases of HL, and the circumstantial evidence is considerable. The incidence of HL is elevated among patients with a history of EBV infection.15, 16, 17 EBV has been associated with other related malignancies, including Burkitt lymphoma and lymphomas developing in immunocompromised patients. Using modern molecular biology techniques, EBV genome fragments have been found in RS cells from approximately 40% of patients with HL,18, 19, 20 more commonly in cases of mixed cellularity HL.23 In addition, the EBV DNA associated with RS cells in HL has been shown to be monoclonal, establishing that EBV preceded the development of HL.18, 19 Interestingly, prognosis appears to be better in HL that is EBV-positive, as compared to cases that are EBV-negative.21, 22, 23
HL occurring in early childhood or in older adults is more likely to be EBV-associated than are cases of HL occurring in young adults.24 However, in an important 2003 study, 38,555 patients with documented EBV-positive infectious mononucleosis were followed for an extensive period.25 In this selected population, 29 cases of HL developed and 16 (55%) were positive for EBV. The risk of EBV-positive HL was increased by a factor of 4 as compared to patients who never had documented EBV-positive infectious mononucleosis; the median latency for HL after EBV infection was 4.1 years. There was no increase of EBV-negative HL in this population. These data do not prove causality: the data are equally compatible with the theory that EBV predisposes patients to the development of HL. If there is a causal role for EBV, additional factors must be required for the development of HL.
The idea that HL may represent an uncommon host response to a common agent has received additional support in a study of monozygotic and dizygotic twins. Monozygotic twins, who would be expected to have similar immune responses, had a 99-fold increased risk of being concordant for having HL, supporting a role for genetic susceptibility or abnormal immune response, or both, in the etiology of HL.26 The incidence of HL in patients with the acquired immunodeficiency syndrome or with human immunodeficiency virus infection is elevated over the incidence in the general population (although not to the extent of the increase in the incidence of non-Hodgkin lymphoma), perhaps lending further plausibility to the concept that HL represents a disordered immune response to antigens.27, 28 An increased incidence of HL has been noted in recipients of allogeneic bone marrow transplants.29 In addition, a patient has been reported with reversible methotrexate-associated lymphoproliferative disorder that eventually evolved into HL.30
HISTOPATHOLOGY
The diagnosis of HL requires biopsy of an involved lymph node or, rarely, of an involved extranodal site (Chapter 86). It is important to note that inflammatory nodes may be interspersed among nodes harboring HL. In general, a larger node with more abnormal features on imaging is a better target for biopsy than a smaller, perhaps more accessible, node. Inguinal nodes, although generally fairly easily biopsied, are often enlarged from current or past inflammation, and biopsy may be misleading. A fine needle aspiration (FNA) should generally be considered an inferior diagnostic approach and will only rarely yield a firm diagnosis; most diagnoses will require an excisional lymph node biopsy (although occasionally a diagnosis can be made on generous core biopsy). Flow cytometry is noninformative in HL, as the majority of cells in a specimen reflect the inflammatory background, with the malignant RS cells being relatively rare. This fact can occasionally falsely reassure a clinician who gets a flow cytometry result of “reactive” and a nondiagnostic FNA on a patient with unexplained adenopathy; the correct response is to go on to an excisional biopsy. Fortunately for clinicians, concordance between pathologists regarding the diagnosis of HL is high, often exceeding 90%.31
HL can be confused with atypical inflammatory reactions that can occur in some patients with infectious mononucleosis32 or in patients receiving phenytoin.33 Among the lymphomas, HL is sometimes mistaken at the histologic level for anaplastic large cell lymphoma, mediastinal large cell lymphoma, or T-cell-rich B-cell lymphoma. Correct classification can usually be achieved using immunohistochemistry and cytogenetic techniques. Recently identified and listed as a provisional entity in the World Health Organizations classification was:34 “B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and classical Hodgkin lymphoma (cHL),” also known as “Grey Zone Lymphoma.”35, 36
Pathologically, HL is distinguished from other lymphomas by the presence of large binucleated or multinucleated cells (RS cells) generally surrounded by a benign reactive response consisting of lymphocytes, histiocytes, granulocytes, eosinophils, and plasma cells. RS cells are large cells with abundant cytoplasm and generally contain two or more nuclei and two or more inclusionlike nucleoli. Molecular evidence (clonal immunoglobulin rearrangements) has shown that the cell of origin for RS cells is a germinal center or post-germinal center B-cell. The RS cell does not express the common B-cell transcription factors and thus its B-cell phenotype (including the B-cell receptor) because of down-regulation and because of epigenetic silencing.37, 38 Commonly such nonfunctional B-cells would undergo apoptosis, and activation of several antiapoptotic pathways (including the nuclear factor [NF]-κB pathway)39 has been proposed as mechanisms for the molecular pathogenesis of HL. The molecular mechanisms involved in the development of HL have been the subject of several reviews,40, 41 and further discussion of these intracellular pathways is beyond the scope of this clinical chapter.
Variant forms of RS cells exist, especially in the nodular sclerosis subtype of HL and the nodular form of lymphocyte-predominant HL (LPHL). RS cells are not specific for HL and have been noted in cases of infectious mononucleosis42 and other malignancies including lymphoma, carcinomas, and sarcomas.43 Therefore, RS cells are not sufficient to establish the diagnosis of HL because that diagnosis depends on the presence of both the characteristic RS cells and the characteristic cellular environment in which the RS cells are found. In addition to the histologic criteria, immunostaining, the RS cells or variants in classical HL should be positive for CD15 (85%) and CD30 (100%), and negative for most pan-B-cell and pan-T-cell antigens. If a B-cell antigen is present, it is usually CD20 and it is variable in intensity.44, 45
The subclassification of HL depends in large part on the ratio of neoplastic to reactive cells and their orientation. The current subtype classification stems from the classification of Lukes and Butler,46 as modified at the Rye Conference in 1966.47 These investigators described RS cell and their variants (Fig. 86.16), one of which they called the L and H cell, (“lymphocytic and histiocytic” because of the associated background) seen primarily in the LPHL. Later these multilobated cells were referred to as “popcorn cells.”48 It was later shown through immunohistochemistry studies that classic RS cells and L and H cells were distinct, with RS cells or variants of other subtypes of HL being CD30 and usually CD15 positive and negative for pan-B-cell markers (Fig. 86.17), and with the L and H cells of LPHL being CD20 pos and CD30 and CD15 negative (Fig. 86.23). Likewise it is now clear on clinical, immunophenotyping, and gene expression profiling grounds that LPHL is a clinicopathologic entity distinct from the other types of HL.49, 50, 51, 52 Thus the current WHO classification34 distinguishes cHL from LPHL.
cHL is comprised of four subtypes: nodular sclerosis HL, mixed cellularity HL, lymphocyte-rich classical HL (LRCHL), and lymphocyte-depleted HL. In the past, the subtype of HL was felt to give the clinician valuable prognostic information; however, it is now recognized that the histologic subtype co-varies with stage, and that much of the reported differences in prognosis previously attributed to histologic subtypes may be attributed to stage. When patients are stratified by stage and receive equivalent therapy, differences attributable to histologic subtype are either trivial or nonexistent.53, 54, 55, 56
The incidence of HL cases with respect to histologic subtype is shown in Table 93.1. The data are taken from one large tumor registry57 and three large series of referred patients,31, 58, 59 and are subject to biases in the selection process. However, the general agreement among the series suggests that this is a fairly reliable estimate regarding the relative incidence of the subtypes of HL.
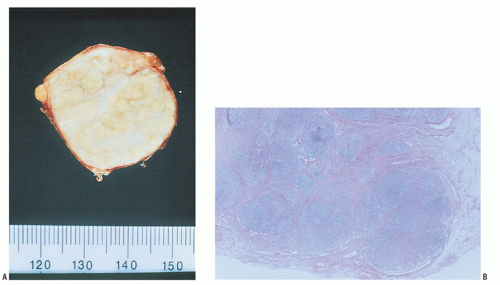
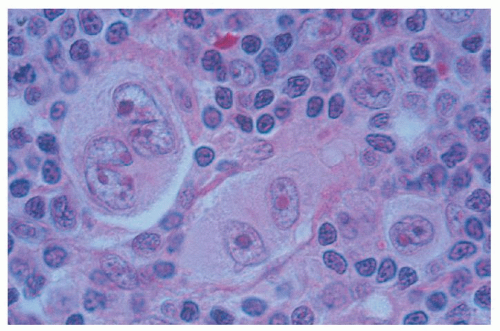
In nodular sclerosis HL, nodularity is produced by dense collagenous bands that divide the cellular portion of the node into sections (Fig. 93.1). In this type of HL, RS cell variants, rather than classic RS cells, are commonly found in the cellular areas. These lacunar RS cells have faintly stained cytoplasm and appear separated from adjacent cells by empty space, an artifact of formalin fixation (Figs. 93.2 and 86.20). Nodular sclerosis HL is the most distinctive form of HL. Although the other subtypes of HL may be regarded as a histologic continuum and transformation among other subtypes is commonly seen, when patients with the nodular sclerosis subtype undergo repeat biopsies, the nodular sclerosis form of HL is confirmed in more than 90% of cases.60 This form of HL classically presents as stage I or II disease with cervical and mediastinal involvement in young adults, although more advanced stages of disease are not uncommon (Table 93.2).
TABLE 93.1 RELATIVE INCIDENCE OF HISTOPATHOLOGIC SUBTYPES OF HODGKIN LYMPHOMA
Note: Percentages refer to “classified” cases, as some studies contain cases of unclassified Hodgkin disease.
A “syncytial variant” of nodular sclerosis HL has been recognized in which numerous RS cell variants have been observed in sheets and clusters. This variant of HL may be confused with NHL, thymoma, or metastatic cancer.61
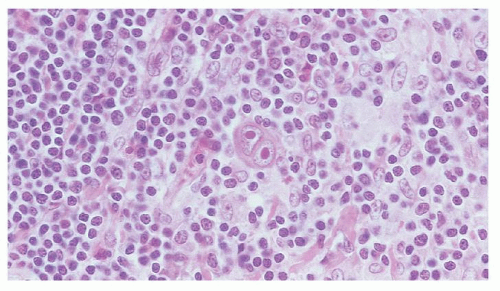
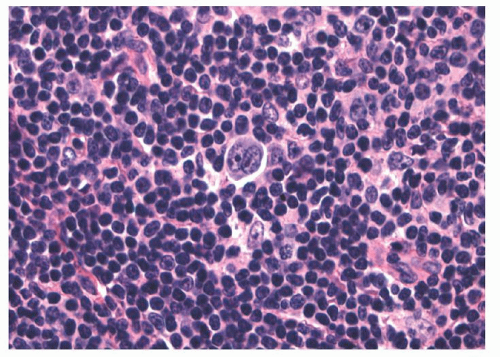
HL of the mixed cellularity pattern is a diffuse lymphoma composed of a mixture of cells including RS cells (Figs. 93.3 and 86.18). Distinguishing mixed cellularity HL from diffuse mixed forms of NHL may be difficult on the basis of histology alone, but should be resolved with modern immunohistochemical stains. This subtype of HL has a greater tendency than nodular sclerosis HL to be advanced at the time of presentation and to be associated with symptomatic disease. Mixed cellularity and nodular sclerosis HL make up the majority of HL cases.
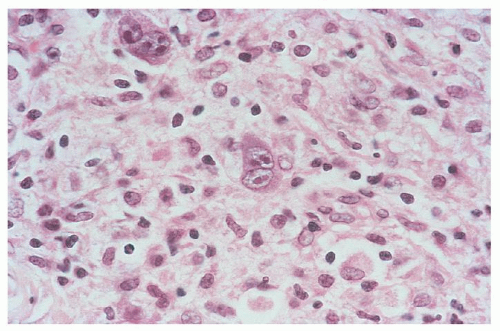
Lymphocyte-depleted HL is composed predominantly of histiocytes and lymphocytes with varying numbers of eosinophils and RS cells (Figs. 93.4 and 86.21). The host reaction is often scant relative to the number of malignant giant cells, and a varying amount of fibrosis is generally present. The disease is the least common type of HL and tends to be advanced at diagnosis (i.e., stage III or IV). Most patients have B symptoms,62 retroperitoneal nodal involvement is common, and approximately one half of patients have bone marrow involvement.63, 64, 65 Many older case series contain cases that would now be classified as peripheral T-cell lymphomas66 or anaplastic large cell lymphomas.67, 68 Therefore the older literature regarding this entirety must be treated cautiously.
FIGURE 93.1. A: Nodular sclerosing Hodgkin lymphoma. Gross appearance of the cut surface of a resected node shows a thickened capsule, white fibrous bands, and yellow parenchymal nodules.B: Nodular sclerosing Hodgkin lymphoma. Low magnification shows a fibrous capsule and bands of sclerosis circumscribing abnormal lymphoid nodules.
LRCHL51 is characterized by rare RS cells or variants dispersed in a background of predominantly small lymphocytes. The tumor cells have the appearance of classic RS cells as well as the immunophenotype of classic RS cells (CD30+, CD15+, but CD20 negative) (Figs. 93.5 and 86.19).
LYMPHOCYTE-PREDOMINANT HODGKIN LYMPHOMA
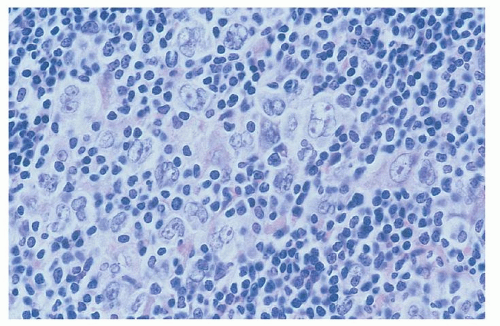
Nodular lymphocyte-predominant HL (NLPHL) is an uncommon subtype of HL, representing about 5% of cases,69 with unique pathologic features distinguishing it from classical HL.70 The neoplastic cell is a large cell, the LP cell, otherwise known as a popcorn cell due to its single, large, folded or multilobulated nucleus that typically has a smaller nucleus than observed in RS cells (Figs. 93.6 and 86.22). However, unlike the RS cell, these cells are typically CD30 and CD15 negative, with CD19, CD20, CD45, and CD79a positivity (Fig. 86.23). These cells are also PAX-5 and Oct-2 positive. The surrounding background lymphocytes are predominantly small CD20 B-cells, with rare eosinophils, neutrophils, or plasma cells. Surrounding the LP cells, CD4+ t-cell rosettes are found and CD21-positive follicular dendritic cells are present, consistent with the germinal center derivation of this malignancy.70
Due to the rare occurrence of this malignancy, presentation, treatment, and patient outcomes are not well described in this disease. In a retrospective analysis of 8,298 patients enrolled on clinical trials for HL through the German Hodgkin Study Group, 394 patients had NLPHL.69 In this series, the median age at diagnosis was 37, 75% patients were male, and 79% of patients had early stage disease. Clinically, there appear to be two age peaks, one in children and another in patients aged 30 to 50. The presence of B-symptoms or bulky disease is unusual, observed in <10% of patients.70 Unlike classical HL, patients with NLPHL typically have peripheral adenopathy (axillary or inguinal) at diagnosis rather than central or mediastinal involvement and nodal spread does not track from LN basin to LN basin as typical for classical HL. In NLPHL, nodal involvement is not contiguous and extranodal involvement is uncommon.
FIGURE 93.2. Nodular sclerosing Hodgkin lymphoma. High magnification shows Reed-Sternberg cells and lacunar variants in B5 fixed material.
An association exists with this subtype of lymphoma and a benign condition, progressive transformation of germinal centers, as well as with NHL, particularly T-cell-rich B-cell lymphoma and diffuse large B-cell lymphoma. Progressive transformation of germinal centers is described as lymph nodes with large, well-defined nodules with an excess of B-cells, or germinal centers overrun by lymphocytes.70 This entity may be observed prior to, simultaneous with, or following a diagnosis of NLPHL. This entity is thought to be a benign condition, but as it occurs concurrently or following a diagnosis of NLPHL, biopsy of recurrent adenopathy is always required in this disease to confirm relapse. Likewise, T-cell-rich B-cell lymphoma and NLPHL can occur simultaneously or in succession, and frequently T-cell-rich B-cell lymphoma can be pathologically confused for NLPHL. With T-cell-rich B-cell lymphoma, large atypical B-cells that are CD20 positive are surrounded by an abundant background of T-cells and histiocytes. As about 5% of NLPHL eventually develop NHL including T-cell-rich B-cell NHL or diffuse large cell lymphoma, biopsy of recurrent lymph nodes is necessary to determine therapy at relapse. In a series of 22 patients treated with rituximab for NLPHL, 9 patients relapsed, including 5 who underwent biopsy at recurrence and of these 2 had diffuse large cell NHL within 13 months of follow-up.71
TABLE 93.2 RELATIVE INCIDENCE OF EACH STAGE FOR EACH HISTOPATHOLOGIC SUBTYPE OF HODGKIN LYMPHOMA
Histologic Subtype
Stage
Lymphocyte Predominant (%)
Nodular Sclerosis (%)
Mixed Cellularity (%)
Lymphocyte Depleted (%)
IA and IB
47
8
12
9
IIA and IIB
38
52
34
14
IIIA and IIIB
14
29
41
41
IVA and IVB
1
11
13
36
Total
100
100
100
100
Modified from Kaplan HS. Hodgkin’s disease, 2nd ed. Cambridge: Harvard University Press, 1980.
FIGURE 93.3. Mixed cellularity-type Hodgkin lymphoma. High magnification shows a classic Reed-Sternberg cell in a mixed background of small lymphocytes, plasma cells, and eosinophils.
CLINICAL EVALUATION
Physical Examination: Sites of Disease
Staging, the basis for treatment planning in HL, begins with the physical examination. HL almost always presents with lymphadenopathy, and the involved nodes are usually freely movable with a rubbery consistency. Cases in which the microscopic appearance reveals fibrosis or sclerosis can be associated with hard firm nodes. Although any lymph node group can be involved (Table 93.3), cervical and supraclavicular adenopathy are the most common physical findings, and axillary presentations are not rare.72, 73
FIGURE 93.4. Lymphocyte-depleted type Hodgkin lymphoma, diffuse fibrosis subtype. Reed-Sternberg cells are easily found, and the background is depleted of cellularity and composed of amorphous eosinophilic connective tissue.
FIGURE 93.5. Lymphocyte-rich “classic” Hodgkin lymphoma. The background is primarily lymphocytes, and the Reed-Sternberg cells are usually CD15+ and CD30+ and negative for the B-cell marker CD20.
Mediastinal disease is often present, but it is rarely the only site of disease because it usually occurs in conjunction with cervical or supraclavicular disease. HL can present with iliac, inguinal, or femoral adenopathy, and in approximately 3% of cases, only subdiaphragmatic disease is present.74, 75 However, in a large series of cases in which the original diagnosis of HL was not confirmed on review, two features that predicted errors in diagnosis were primary extranodal disease and primary subdiaphragmatic disease.76 Thus, the pathologic diagnosis of HL should be questioned in cases with subdiaphragmatic presentation. A complete physical examination of the patient with HL should include an evaluation of Waldeyer’s ring and epitrochlear nodes even though these sites are only rarely involved.
Splenomegaly is noted at presentation in approximately 10% of cases of HL. However, splenomegaly may be a nonspecific manifestation of the HL, and in only one half of patients with splenomegaly was splenic involvement confirmed at laparotomy73 in a study from the era of staging laparotomies. Additionally, splenic involvement may occur in 20% to 30% of patients in the absence of splenomegaly73 even when abdominal computed tomography (CT) scans are used to assess splenomegaly by three-dimensional measurements.77 This latter fact should not be surprising, because splenic involvement may be limited to a few microscopic nodules.
TABLE 93.3 FREQUENCY OF INVOLVEMENT OF NODAL SITES IN HODGKIN LYMPHOMA
Site
Frequency of Involvement (%)
Frequency of Involvement as Only Site of Disease (%)
Modified from Kaplan HS. Contiguity and progression in Hodgkin’s disease. Cancer Res 1971;31:1811-1813; and Kaplan HS, Dorfman RF, Nelsen TS, et al. Staging laparotomy and splenectomy in Hodgkin’s disease: analysis of indications and patterns of involvement in 285 consecutive, unselected patients. Natl Cancer Inst Monogr 1973;36:291—301.
FIGURE 93.6. Lymphocyte-predominant Hodgkin lymphoma. High magnification shows variant lymphocytic and histiocytic cells (L and H cells), which have “popcorn” nuclei. A background of small lymphocytes and histiocytes is present.
Extranodal disease can occur at any site. However, the central nervous system and the testis, which are not uncommon sites for NHL, are exceptionally rare sites for HL. At presentation, lung, liver, bone, and bone marrow are the most common extranodal sites of disease, with each of these sites seen in approximately 5% to 10% of cases.78, 79 Disease at other individual sites occurs in <5% of cases. Primary extranodal HL should always lead the clinician to consider the possibility of an incorrect pathologic diagnosis.
Pattern of Spread
HL tends to spread in a contiguous fashion.72, 80 In fact, when one considers that the left supraclavicular area and the upper abdomen are contiguous (via the thoracic duct), a contiguous pattern of disease can be established at presentation in more than 90% of cases.72 For patients with only right supraclavicular involvement at presentation, abdominal disease is found in only 8% of cases; for patients with left supraclavicular involvement at presentation, the incidence of abdominal involvement is 40%. Hilar involvement with HL does not occur unless mediastinal disease is present. Pulmonary involvement by HL does not occur in the absence of mediastinal and hilar involvement.81
The spleen is the most commonly involved abdominal site of disease, possibly representing hematogenous spread. Liver involvement is uncommon at presentation in patients with HL, and it is exceptionally rare in the absence of splenic involvement.72, 82 Within the abdomen, the pattern of disease detected at staging laparotomy suggests spread from the spleen in a contiguous fashion. In a series in which staging laparotomies were performed with rigorous attention to biopsy of all nodal groups, the spleen and contiguous nodes were involved in 71% of cases, the spleen and noncontiguous nodes were involved in 16% of cases, and lymph nodes were involved in the absence of splenomegaly in only 13% of cases.83 It is unknown whether the cases of apparent discontiguous spread represent cases in which early hematogenous dissemination has occurred or cases in which HL has spread through a node without involving the node.
Initial Evaluation of the Hodgkin Lymphoma Patient
Clinical History
An accurate clinical history facilitates the management of HL patients in a number of ways. The presence of certain symptoms is associated with a less favorable prognosis and may be a clue to more advanced disease than otherwise suspected. Other symptoms may point out the need for additional studies (such as a bone scan or neck CT) for accurate staging. The discovery of additional sites of disease because of symptoms may lead to a modification of therapy and establish the need for additional follow-up studies on completion of treatment. A history of heart disease or lung disease may prompt closer assessment of those organs before chemotherapy or during its course. A history of HIV infection may affect choice of chemotherapy or infection prophylaxis. A history of smoking or autoimmune disease or a family history of breast cancer may influence the decision to incorporate radiation therapy into the treatment plan and may influence the follow-up plan.
The constitutional symptoms that are known to have prognostic value are unexplained fever to >101°F, drenching night sweats, and weight loss equal to 10% of the patient’s weight. These are the only symptoms that are accounted for in the Cotswold staging system (and whose presence is denoted by B appended on the numerical stage). Fever in HL can have any pattern, including continuous low-grade fever or occasional fever spikes. The pattern of recurrent episodes of daily high fevers separated by days without fever, Pel-Ebstein fever, was first associated with HL in 1885.84, 85 However, in the modern era it is a rare manifestation of HL and, although limited to patients with very advanced disease, it is uncommon even in those patients. Rarely, fever may be the only clinical manifestation of HL, for instance, in the case of patients with the lymphocyte-depleted HL who present with fever and disease limited to retroperitoneal nodes or the bone marrow, or both.86
Before designating a patient as having the symptom of night sweats, the clinician should verify that the patient’s sweating is abnormal. The night sweats of HL are drenching and not simply associated with increased ambient temperature. Perimenopausal women should be asked if they have vasomotor symptoms before the sweating, as flushing is not generally a symptom of the sweats of HL.
Pain at the site of HL in association with the ingestion of alcohol is well described and can be the first hint of a recurrence. The mechanism of this phenomenon is unknown and it does not have prognostic significance. The occurrence of alcohol-induced pain has become less common in recent decades, perhaps in association with earlier diagnosis and more effective therapy. Generalized pruritus may be a nonspecific manifestation of HL, and it may be an early manifestation of the disease in up to 10% to 15% patients. It also has no known prognostic implication. Similarly, fatigue and weakness may occur in patients with HL but do not cause a patient to be classified as symptomatic.
Although the staging classification divides patients into categories A and B for asymptomatic and symptomatic, respectively, clinicians are well aware that this binary classification is an oversimplification and that the presence 0 of symptoms varies along a continuum87, 88 with respect to their clinical significance. When patients with stage IB and stage IIB HL were reviewed, it was found that the presence of fevers was associated with a relative risk of relapse after treatment of 4.3. For weight loss alone, the relative risk of relapse after treatment was 2.4; for night sweats, the relative risk of relapse was 0.8 (i.e., not increased).87 Nevertheless, the presence of symptoms may suggest advanced disease. Symptoms are found in <10% of patients with stage I disease and in approximately two thirds of patients with stage IV disease.89 The presence of B symptoms in a patient with apparent stage I disease is, therefore, suggestive that further evaluation will lead to the discovery of a more advanced-stage disease.
The Cotswold Staging System
The stage of the patient is the main determinant of therapy and prognosis in HL. The current classification system is the Cotswold classification system90, 91 (Table 93.4). Although the modalities used to stage patients have changed since the Cotswold conference in 1990 (e.g., PET and improved CTs have replaced staging laparotomy and gallium scans), and treatment algorithms have changed, the current staging system remains the backbone for clinical decision making and for clinical trial design.
Stage I disease is the involvement of a single lymph node region (or a single lymphoid structure). Stage II disease is the involvement of multiple lymph node regions (e.g., cervical nodes, supraclavicular nodes) but on only one side of the diaphragm. Stage III disease is the involvement of lymph node regions on both sides of the diaphragm and is further subdivided as to anatomic substage. Disease limited to the upper abdomen (i.e., spleen, splenic hilar nodes, celiac nodes, or porta hepatis nodes) in a patient with stage III disease is defined as substage III1. Patients with stage III disease in whom abdominal nodal involvement includes para-aortic, iliac, or inguinal nodes are classified as substage III2. The distinction between stages III1 and III2 is largely obsolete as all patients with stage III are now treated similarly with chemotherapy. Stage IV disease is visceral involvement (e.g., lung, liver, or bone marrow) that is not due to direct extension from a nodal site. The subscript E represents extension from a nodal site, such as extension of a mediastinal mass directly into the lung, for example, IIE.78 The designations A and B are used to represent the absence and presence of symptoms (e.g., unexplained fever, drenching night sweats, and weight loss of 10% of body weight), respectively. The presence of “bulky” disease, defined as a mediastinal mass with a diameter greater than one-third the diameter of the chest as measured at the T5-T6 interspace on a postero-anterior chest radiograph, or any nodal mass with a greatest diameter of more than 10 cm, is represented by the designation X.92, 93, 94 The relative incidence of each stage of HL is shown in Table 93.5.
TABLE 93.4 COTSWOLD STAGING CLASSIFICATION FOR HODGKIN LYMPHOMA
Stage I
Involvement of a single lymph node region or lymphoid structure (e.g., spleen, thymus, Waldeyer ring)
Stage II
Involvement of two or more lymph node regions on the same side of the diaphragm (the mediastinum is a single site; hilar lymph nodes are lateralized); number of anatomic sites should be indicated by a suffix (e.g., II3)
Stage III
Involvement of lymph node regions of structures on both sides of the diaphragm
III1
With or without splenic hilar, celiac, or portal nodes
III2
With para-aortic, iliac, or mesenteric nodes
Stage IV
Involvement of extranodal site(s) beyond that designated E
A
No symptoms
B
Fever, drenching sweats, weight loss
X
Bulky disease, greater than one-third widening of the mediastinum, >10 cm maximum dimension of nodal mass
E
Involvement of a single extranodal site, contiguous or proximal to a known nodal site
CS
Clinical stage
PS
Pathologic stage
TABLE 93.5 DISTRIBUTION OF HODGKIN LYMPHOMA CASES WITH RESPECT TO STAGE AND SYMPTOMATIC STATUS
Stage
A (%)
B (%)
A and B (%)
I
11.4
0.8
12.2
II
34.1
12.4
46.5
III
17.5
13.1
30.6
IV
3.3
7.4
10.7
Total
66.3
33.7
100.0
Modified from Kaplan HS. Hodgkin’s disease, 2nd ed. Cambridge: Harvard University Press, 1980.
Required and Suggested Studies for Initial Evaluation
As treatment recommendations for HL depend heavily on stage, accurate staging is crucial. Staging also establishes a baseline so that the completeness of remission can be assessed on completion of therapy. The evaluation begins with a clinical history (see above) and a physical examination. The physical exam should focus on accessible lymphoid tissues, including Waldeyer’s ring, all palpable lymph node areas, including the epitrochlear and popliteal nodes, as well as the spleen.
The role of radiographic imaging in HL patients is to determine the stage accurately, as stage determines treatment. A second role is to establish a baseline for comparison to mid-treatment or post-treatment scans. There is now a general agreement that positron emission tomography with integrated CT scanning (PET/CT) can supplant other imaging studies in the initial assessment of most patients with HL. PET/CT is reported to have very high sensitivity and specificity in HL. CT alone relies on size criteria to distinguish normal nodes from involved nodes and thus may understage patients with small but involved nodes. It is also poor at identifying a diffusely involved organ such as the spleen or liver, unless those organs are enlarged. It is reported that PET/CT (as compared to CT alone) changes the stage in up to 20% to 40% of cases and changes the treatment in 5% to 15%.95, 96, 97 The majority of these cases are upstaging. Some argue for omitting PET/CT in favor of CT alone for certain “clearly limited-stage” patients,98 however, most clinicians and most guidelines (e.g., the National Comprehensive Cancer Network) favor initial PET/CT as a routine study for initial evaluation of HL. If a PET/CT is unavailable, or otherwise unused, patients should have CT scans of the chest, abdomen, and pelvis. A neck CT should be included if there is any clinical suspicion of involvement of neck nodes. A chest radiograph is performed in a patient with a mediastinal mass to assess whether the patient’s disease meets the criteria for bulky mediastinal disease. Certain situations and patients may warrant other radiographic studies, for example, the patient with bone pain may be evaluated with bone scan or MRI.
A marrow biopsy is usually performed on any patient with B symptoms or abnormal blood counts, and on any patient with stage III or IV disease. Although the chance that an early stage asymptomatic patient with normal blood counts will have marrow involvement is low,79, 99 some clinicians perform a marrow biopsy on all patients with HL. Recent data suggest that PET/CT accurately identifies or rules out marrow involvement,100 and clinicians may be able to decrease their use of marrow biopsy accordingly in the future.
Blood tests should include a complete blood count with differential and platelets, erythrocyte sedimentation rate, liver function tests including alkaline phosphatase, lactate dehydrogenase, albumin and bilirubin (abnormal liver function tests are not diagnostic of liver involvement, but are sometimes nonspecifically elevated and may complicate the use of certain chemotherapy agents); BUN and creatinine, and thyroid function tests as a baseline (if neck radiotherapy is contemplated). Serology for human immunodeficiency virus is appropriate in selected cases. A pregnancy test should be performed for women of childbearing potential.
Patients should be counseled about fertility preservation. An assessment of ejection fraction by echocardiogram or radionuclide study is appropriate if anthracycline chemotherapy will be used. Pulmonary function tests with diffusing capacity should be ordered if bleomycin-containing chemotherapy will be used.
A summary list of suggested initial procedures and tests is presented in Table 93.6.
Prognostic Indicators
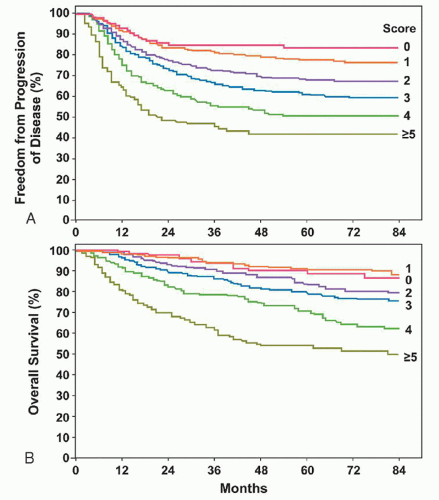
As noted above, stage is the major prognostic factor in HL and the major basis on which therapy is determined. However, patients can be stratified for risk on the basis of prognostic factors in addition to stage. In a study of 5,141 patients with “advanced” HL, primarily stage III and IV disease, Hasenclever and Diehl101 performed a multivariate analysis of risk factors (Fig. 93.7). Seven independent factors that predicted freedom from progression (FFP) were identified: albumin <4.0 g/dl; hemoglobin <10.5 g/dl; male sex; 45 years of age or more; stage IV disease; leukocytosis at or above 15,000/mm3; and lymphocytopenia (lymphocytes ≤600/mm3 and/or lymphocytes <8% of total white count). For patients with no risk factors (7% of all patients), FFP was 84%. For patients with one risk factor (22% of all patients), FFP was 77%. For patients with two risk factors (29% of all patients), FFP was 67%. For patients with three risk factors (23% of all patients), FFP was 60%. For patients with four risk factors (12% of all patients), FFP was 51%. For patients with five or more risk factors (7% of all patients), FFP was 42%. Of note, B symptoms did not have independent prognostic value in this model. At present this model is validated only for advanced-stage patients.
TABLE 93.6 RECOMMENDED INITIAL EVALUATION FOR PATIENTS WITH HODGKIN LYMPHOMA
History and Physical Examination, Including
Past medical history: including B symptoms (fever, night sweats, weight loss of >10% in past 6 mo), alcohol intolerance, itching, HIV risks, cardiac, renal, liver impairment
Family history: including lung cancer, coronary disease, breast cancer
Personal history: smoking
Examination of all peripheral lymph node regions (including epitrochlear, popliteal, Waldeyer’s ring), liver, and spleen
Radiologic Studies
Chest radiograph
PET/CT
Assessment of EF (optional in younger patients)
Pulmonary function tests (optional in younger patients)
Laboratory Studies
Hematocrit, white blood cell count, differential, platelet count
FIGURE 93.7. Progression-free survival (A) and survival (B) as related to number of risk factors. (From Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. N Engl J Med 1998;339:1506-1514, with permission.)
A number of prognostic classifications have been used for early-stage HL and have been used to tailor therapy further in clinical trials. These commonly employ some combination of age, erythrocyte sedimentation rate, mediastinal bulk, and number of involved nodal sites. See Table 93.7, “Unfavorable Prognostic Factors for Stage I—II HL, by Three Cooperative Groups.”
OVERVIEW AND HISTORICAL PERSPECTIVE OF RADIATION THERAPY
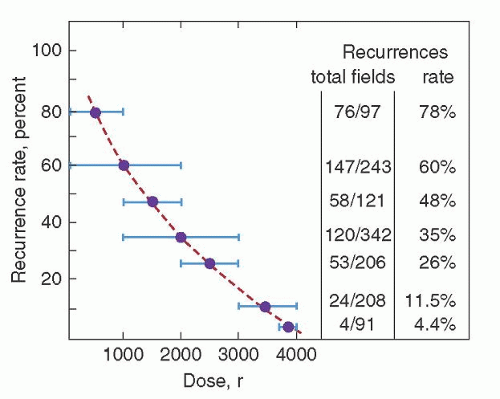
Six years after the discovery of x-rays by Roentgen in 1895, Pusey reported that x-rays could shrink enlarged nodes in patients with HL.102 However, given the orthovoltage techniques of the day, therapy was only palliative. The modern radiotherapy era began with the Swiss radiotherapist Gilbert in 1925.103 Based on observed patterns of spread in patients with HL, Gilbert advocated treatment of both involved areas and adjacent, apparently uninvolved, areas. This approach was also adopted by Peters, who, in 1950, was the first to report that radiation therapy of HL could produce cures.104 Over the next two decades, the curability of HL was confirmed by Peters,105, 106 Easson and Russell,107 and Kaplan,108, 109 the latter investigator establishing the critical relationship between radiation dose and the risk of recurrence in the treatment field (Fig. 93.8).
TABLE 93.7 UNFAVORABLE CHARACTERISTICS FOR STAGE I AND II HL, PER THREE COOPERATIVE GROUPS
Risk Factor
GHSG
EORTC
NCIC
Age
≥50
≥40
Histology
NC or LD
ESR and B symptoms
>50 if A; >30 if B
>50 if A; >30 if B
50 or any B
Mediastinal mass
MMR > .33
MTR > 35
MMR >.33 or >10 cm
# Nodal sites
>2a
>3
>3
E lesions
any
GSHG, German Hodgkin Study Group (auses alternate definition of nodal sites); EORTC, European Organization for the Research and Treatment of Cancer; NCIC, National Cancer institute of Canada; MMR, mediastinal mass ratio: maximal width of mass/maximal intrathoracic width; MTR, mass to thoracic width at T5-T6 interspace on standing PA chest radiograph.
As originally suggested by Gilbert, the principle of treating involved and adjacent apparently uninvolved areas (i.e., extended field therapy) became a standard radiotherapeutic approach to HL. With the advent of staging laparotomy and evidence that the retroperitoneum could represent a potentially involved adjacent area of disease, the concept of extended field therapy came to include the standard mantle, para-aortic/splenic, and pelvic radiation ports shown in Figure 93.9.110
Although the radiotherapy pioneers were the first to cure HL, and they did so by conducting elegant and groundbreaking clinical trials, the era of radiation as single modality for the initial treatment of HL has passed. However, HL is a very radiation-sensitive disease, and radiation remains a useful component of the treatment of certain patients. Currently the most well documented role for radiation is in the early stage setting, discussed below.
Complications of radiation therapy depend on technique, dose, and the volume of irradiated tissue. Common complications include hair loss within the treatment fields, radiation pneumonitis, radiation pericarditis, mediastinal fibrosis, and pulmonary fibrosis. Symptoms of radiation pneumonitis generally occur within 1 to 3 months of completing therapy and are nonspecific because they include dyspnea, cough, and fever. Such complications are rarely seen at lung doses <15 Gy and occurred in <5% of patients receiving routine mantle irradiation. The presence of a large mediastinal mass or the concomitant use of chemotherapy essentially doubles the risk of radiation pneumonitis.111, 112 Although infection and recurrent HL must be considered in the differential diagnosis of radiation pneumonitis, radiation injury is likely to be confined to the area of the lung that was irradiated. Infiltrates with sharp borders, representing the edge of the radiation field, strongly suggest the presence of radiation pneumonitis. Because HL and its therapy are both associated with immunosuppression, whenever radiation injury is considered in the differential diagnosis of a patient with HL, one must also consider the diagnosis of Pneumocystis jiroveci pneumonia.
FIGURE 93.8. Rate of recurrence in a given treatment field as a function of radiation dose delivered to that field. (From Kaplan HS. Evidence for a tumoricidal dose level in the radiotherapy of Hodgkin disease. Cancer Res 1966;26:1221-1224, with permission.)
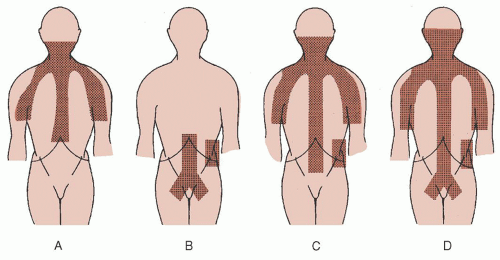
FIGURE 93.9. Treatment fields used as extended field irradiation of Hodgkin disease. A: Mantle field. B: Inverted Y field. C: Mantle and para-aortic field (extended mantle field). D: Total nodal field. The spleen is irradiated in conjunction with the fields in B, C, and D, unless it has been surgically removed.
Cardiac toxicity of radiation therapy can include myocarditis, arrhythmias, pericarditis (rarely including constrictive pericarditis and tamponade), valvular heart disease, and coronary artery disease.113, 114, 115, 116, 117, 118 Although an increased risk of myocardial infarction appears to exist after radiation therapy,117 this risk is greater for patients who received radiation therapy before 1966, suggesting that modern treatment techniques have minimized this risk.
Spinal cord injury should not occur with modern radiation therapy techniques but may occur if radiation fields overlap. Splicing of fields over the spinal cord should be done with great care. The one neurologic problem that may occur despite the use of appropriate technique is the Lhermitte sign,89 which includes numbness and tingling in the arms, legs, or both and electrical sensations up and down the spine made worse by flexion of the head. This complication is usually transient and of no clinical consequence.
Hypothyroidism is a common complication of radiation therapy,119 with actuarial analysis indicating that 52% of patients develop hypothyroidism 20 years after radiation.119 Thyroid cancer is an uncommon complication, but, based on the hypothesis that chronic elevations of thyroid-stimulating hormone in patients after thyroid irradiation may lead to thyroid tumors, the administration of thyroid replacement has been advocated in patients with an elevated thyroid-stimulating hormone even if they are clinically euthyroid.119
In addition to thyroid cancer, an increased risk of second malignancies, including lung cancer,120, 121 stomach cancer,120 melanoma,120, 122 and breast cancer,123, 124, 125 has been noted in patients receiving radiation therapy for HL. These malignancies generally occur 10 to 20 years after radiation therapy, with the risk of second malignancies being increased in patients who have also received chemotherapy. The relative risk of breast cancer secondary to radiation is age related. The relative risk of developing breast cancer is 38 for patients radiated before age 20; the relative risk is 17 for patients radiated between the ages of 20 and 29; the relative risk is 4 for patients radiated after the age of 30, a relative risk that is not significantly different from 1.123 Fortunately, the baseline risk of breast cancer in patients younger than age 30 is low, muting the quantitative impact of this increase in relative risk. Nevertheless, it has been recommended that women who receive radiation therapy for HL should receive mammography starting 8 years after completion of therapy because 95% of such tumors occur more than 10 years after radiation.124
Only gold members can continue reading. Log In or Register to continue