II. PATHOLOGY AND NATURAL HISTORY
A. Histology. The pathologic diagnosis of HL depends on the presence of RS cells and their variants in
an appropriate pathologic milieu. The bulk of lymphatic tissue involved by HL is not composed of neoplastic cells but rather a variety of normal-appearing lymphocytes, plasma cells, eosinophils, neutrophils, and histiocytes existing in different proportions in the various histologic subtypes. Important variants of RS cells include L&H (lymphocyte and histiocyte) cells, lacunar cells, and RS-like cells (see
Appendix C5. Discriminatory Immunophenotypes for Lymphocytic Neoplasms).
1. The Rye classification for HL relates the histopathologic subtypes to clinical behavior and prognosis. This older classification system comprises lymphocyte-predominant (LP), nodular sclerosing (NS), mixed-cellularity (MC), and the uncommon lymphocyte-depleted (LD) varieties of HL. The LP subtype was further divided into nodular LP and diffuse LP subtypes. Immunohistochemistry, however, has resulted in deletion of the diffuse LP subtype and redefinition of the nodular LP subtype (see Section II.A.2).
2. The World Health Organization (WHO) classification divides HL into nodular LP HL and classical HL. Classical HL in this newer classification system comprises the lymphocyte-rich, NS, MC, and LD varieties.
a. Nodular LP HL with its L&H cells, which are not classical RS cells, is now clearly recognized to be most like an indolent B-cell NHL and not true HL. For that reason, nodular LP HL is distinguished from classical HL in the WHO classification.
Table 21.2 shows this classification system with distinguishing histopathologic features, clinical correlates, and immunophenotypes.
b. Diffuse LP HL in the Rye classification has disappeared as an entity. In the new WHO classification of lymphocytic neoplasms (see
Appendix C6), what was thought to be diffuse LP HL is now classified as lymphocyte-rich classical HL (with true RS cells that are CD30-positive), Lennert lymphoma (lymphoepithelioid peripheral T-cell lymphoma), T-cell-rich B-cell lymphoma, or other entities.
3. RS cells and their variants
a. RS cells are giant cells that have more than one nucleus and large, eosinophilic, inclusion-like nuclei. Single-cell polymerase chain reaction analysis has shown that the RS cells are B cells that originate in the germinal centers of lymph nodes. RS cells and the accompanying mononuclear
Hodgkin cell variants are the neoplastic cells in HL and are surrounded by a reactive cellular infiltrate. Classic RS cells usually express CD15
and CD30. CD30 (Ki-1) is an antigen that is also expressed in anaplastic large cell lymphoma and occasionally in other forms of NHL (e.g., large B-cell lymphoma [LBCL], peripheral T-cell lymphoma [PTCL]). RS cells express CD20 infrequently, but not CD45 (leukocyte common antigen).
b. The lacunar cell is a variant of the RS cell and has the same immunophenotype. It characterizes NS HL and is often far more plentiful than classic RS cells in that subtype.
c. L&H cells are RS-like but have a different immunophenotype. L&H cells manifest B-cell markers (CD20, CD45, and CD79a), but not CD15 or CD30. Although the L&H cells are believed to be of monoclonal origin, the surrounding B-cell infiltrates may be polyclonal. L&H cells were identified in nodular LP HL, which is now considered a separate entity because of its distinct immunophenotype.
d. RS-like cells are found in a variety of infectious, inflammatory, and neoplastic disorders, including infectious mononucleosis, lymphoid hyperplasia associated with phenytoin therapy, and immunoblastic lymphomas.
B. Mode of spread (see
Table 21.1). HL almost always originates in a lymph node. Whenever a primary diagnosis of HL is made in an extranodal site without contiguous nodal involvement, the diagnosis should be highly suspect. For much of its natural history, HL appears to spread in an orderly fashion through the lymphatic system by contiguity. Histologic types other than NS, however, often skip the mediastinum, and disease appears in the neck and upper abdomen. The axial lymphatic system is almost always affected in HL, whereas distal sites (e.g., epitrochlear and popliteal nodes) are rarely involved. Hematogenous dissemination occurs late in the course of disease and is characteristic of the LD subtype.
C. Sites of involvement
1. Peripheral lymph nodes. Cervical or supraclavicular lymphadenopathy occurs in >70% of cases. Axillary and inguinal lymph nodes are less frequently involved. Generalized lymphadenopathy is atypical of HL. Left supraclavicular lymphadenopathy is more strongly associated with abdominal involvement (specifically, splenic involvement) than is right-sided disease.
2. Thorax
a. The anterior mediastinum is a prime location for NS HL. Mediastinal precedes hilar lymph node involvement.
b. Lung involvement may occur by direct contiguity with hilar involvement in HL as well as by hematogenous dissemination. Pulmonary involvement by HL may produce discrete nodules and irregular, interstitial, or even lobar infiltrates.
c. Pleural effusion may occur secondary to mediastinal compression of vascular-lymphatic drainage and by direct pleural involvement. Chylous effusions occasionally occur.
d. Pericardial involvement may be found on CT scans, but overt cardiac tamponade is uncommon.
e. Superior vena cava syndrome is more frequent in NHL than in HL.
3. Spleen, liver, and upper abdomen
a. The spleen, splenic hilar nodes, and celiac nodes are the earliest abdominal sites of involvement in infradiaphragmatic HL. Mesenteric lymph nodes are rarely involved in HL.
b. At least 25% of spleens not clinically enlarged harbor occult HL at laparotomy, and as many as half of spleens believed to be enlarged on physical examination or radiologic assessment are histologically normal.
c. Liver involvement is uncommon at diagnosis and is almost always associated with infiltration of the spleen.
4. Retroperitoneal lymph node involvement tends to occur relatively late in the course of supradiaphragmatic HL and after spleen, splenic hilar, and celiac nodal involvement. Periaortic involvement without splenic involvement is uncommon. The retroperitoneal nodes are, however, affected early in the course of inguinal presentations of HL.
5. The bone marrow is rarely involved at the time of diagnosis of HL. Patients with advanced-stage disease, systemic symptoms, and MC or LD histologies have a higher risk for bone marrow involvement. Biopsy is mandatory to evaluate the bone marrow because HL is difficult to diagnose on marrow aspirates. Granulomatous changes or fibrosis may be seen, which is not diagnostic of HL involvement.
6. Bone. Osseous involvement of HL usually produces an osteoblastic reaction mimicking prostatic carcinoma. Extradural masses may result in spinal cord compression. Sternal erosion by mediastinal NS HL may occur.
7. Other extranodal sites are rarely involved in HL. Liver or skin involvement is rare and usually a late manifestation of disease. CNS involvement is very uncommon with the exception of extrinsic spinal cord compression. Clinical involvement of meninges, brain, Waldeyer ring, GI tract, kidney, and other extranodal sites usually suggests an alternative diagnosis.
III. STAGING SYSTEM AND PROGNOSTIC FACTORS
A. Staging is the most crucial determinant of prognosis and treatment in HL. The
Ann Arbor Staging System had previously been universally used but has been modified to take into account important prognostic factors, particularly mediastinal bulk. The modified system is called the
Cotswolds Staging Classification and is shown in
Table 21.3.
B. Prognostic factors
1. Stage is clearly the single most important prognostic factor in HL. Within each stage, the presence of B symptoms confers a poorer prognosis. About 60% of patients with HL in the United States have stage I or II disease at the time of diagnosis. The percentage of patients with stage III or IV disease is generally higher in Third World countries and in lower socioeconomic enclaves.
2. Histopathology. With advances in therapy, the value of histopathologic subtype as an independent prognostic variable (apart from stage) is less clearly defined than it was in the past.
3. Adverse prognostic factors were evaluated by an international group in a multivariate retrospective analysis of 4,695 patients, mostly with extensive disease (see
Hasenclever et al. 1998 in “Suggested Reading”). Patients with no adverse factors had an 84% freedom from progression, whereas the presence of
each factor depressed the freedom from progression curve plateau by about 8%. Interestingly, neither tumor bulk nor histology emerged as independent factors. The seven independent prognostic factors identified were as follows:
4. Independent adverse prognostic factors for NS HL include eosinophilia, lymphocyte depletion, and RS cell atypia.
5. Adverse prognostic factors in early stage HL include ESR ≥50 mm/h, four or more separate sites of nodal involvement, bulky mediastinal mass (defined as >33% of the maximum intrathoracic diameter) or any mass ≥10 cm, or extranodal sites of disease.
V. MANAGEMENT: PRIMARY THERAPY
A. Treatment philosophy. More than one treatment approach may be used in the management of cases of HL. The challenge is to determine a course of therapy that preserves cure while minimizing long-term complications.
B. Surgery is limited to diagnosis, possibly laparotomy, and laminectomy for spinal cord compression.
C. RT alone is still used in the United States to treat many patients with stage IA or possibly IIA disease nonclassical HL. However, it is increasingly replaced in the treatment of classical HL with combined-modality treatment.
1. Radiation dose. HL may be locally sterilized in almost all cases with 3,000 to 4,400 cGy given at a rate of about 1,000 cGy per week. Lesser doses may be adequate as consolidation after chemotherapy.
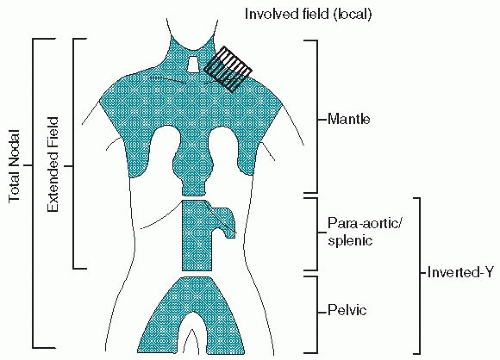
a. Mantle field encompasses the cervical, supraclavicular, infraclavicular, axillary, hilar, and mediastinal lymph nodes to the level of the diaphragm. Preauricular fields are added for patients with high cervical lymphadenopathy. The lungs and much of the heart are shielded by lead blocks, although many radiotherapists administer some radiation (≤1,500 cGy) to the lung on the involved side, if hilar lymph nodes are enlarged. The whole heart may be treated if the pericardium is involved. A small gap must be left between the inferior border of the mantle field and the superior border of the periaortic field to obviate potential severe spinal cord injury caused by overlap.
b. Inverted-Y field includes the spleen or splenic pedicle and the celiac, periaortic, iliac, inguinal, and femoral lymph nodes. The kidneys, much of the pelvic marrow, and the testes are shielded.
c. Spade and pelvic fields. The inverted-Y field may be divided into a spade field, encompassing the splenic pedicle (or spleen) and periaortic nodes, and a pelvic field, including the iliac, inguinal, and femoral lymph nodes.
d. Subtotal nodal or subtotal lymphoid irradiation consists of mantle and spade fields.
e. Total nodal or total lymphoid irradiation is uncommonly used and consists of mantle and inverted-Y fields.
f. Involved-field radiation therapy (IFRT) consists of sites of known disease only and is used with curative intent only in combination with chemotherapy. It has become the most common use of RT in HL with other fields previously described of mostly historical interest. Doses given in combined-modality therapy range from 2,000 to 3,600 cGy.
D. Combination chemotherapy is the mainstay modality for all stages of classical HL and advanced stages of nonclassical HL. Chemotherapy, often in combination with RT, is also preferable for patients with early-stage disease and/or bulky disease. The selection among the available regimens is often guided by the desire to avoid long-term toxicities associated with specific treatments. The advent of the nonleukemogenic, gonadal-sparing ABVD chemotherapy regimen expanded the use of chemotherapy to patients with earlier stages and obviated the need for laparotomy; it has replaced the historic MOPP. More aggressive regimens such as BEACOPP (
Appendix D1, Section II) may improve on ABVD (see Section V.D.3), especially in patients with advanced disease. Maintenance therapy is not recommended.
1. Useful chemotherapy regimens for HL are shown in
Appendix D1. These regimens must be strictly followed because delays in therapy or reduction in dosages not indicated by the protocol can clearly compromise results. The total dose and dose rate (dose intensity) are important in achieving cure. Regimens used as salvage therapy in HL are shown in
Appendix D3.
2. MOPP or COPP regimen (
Appendix D1, Section I). The National Cancer Institute (NCI) recommends that vincristine should not be limited to a 2-mg maximum dosage in this regimen, but most clinicians sustain the 2-mg limit. Treatment is administered in 28-day cycles for two additional cycles beyond the attainment of a restaged complete response (CR) and a minimum of six cycles (6 months).
a. The CR rate using the MOPP regimen is between 70% and 80% for stages III and IV HL. About 60% to 70% of CR cases are durable, with relapses rare after 42 months. More than 80% of patients with stage IIIA or IVA disease survive 10 years without recurrence of disease. Histologic subtype appears to have little effect on results with MOPP.
b. The MOPP regimen is particularly emetogenic, and it is associated with myelosuppression neuropathy, leukemogenesis, and infertility. It is believed that COPP (replacing mechlorethamine with cyclophosphamide) may be better tolerated.
3. ABVD regimen (
Appendix D1, Section I) is superior to the MOPP regimen and causes much less leukemia and infertility. Potential cardiac toxicity caused by doxorubicin and pulmonary toxicity caused by bleomycin (particularly with the concomitant use of granulocyte growth factors) have been occasional problems using this schedule. The concern is heightened when combined with mediastinal RT. ABVD-based treatment has replaced MOPP as the standard regimen for HL.
a. Generally, the same therapeutic rules as with the MOPP regimen apply: 6 to 8 monthly cycles are usually administered and at least two cycles beyond maximum response.
b. Pulmonary function should be monitored. If dyspnea, pneumonitis, or significant reduction to <40% of predicted lung diffusion capacity is noted, bleomycin should be discontinued. Bleomycin pneumonitis usually responds to corticosteroids and mandates discontinuation of bleomycin. As there is a concern that the use of myeloid growth factors may increase the risk for bleomycin pulmonary toxicity, their use has been discouraged.
c. Cardiac function should be monitored in patients with pre-existing heart disease and in those receiving high cumulative doses of doxorubicin. A baseline measurement of left ventricular ejection fraction is suggested before beginning doxorubicin administration.
4. MOPP and ABVD in alternating cycles and the MOPP/ABV hybrid have both been found to be less satisfactory than ABVD alone. While MOPP/ABV and ABVD were equally efficacious, the hybrid regimen was associated with increased acute toxicity, myelodysplastic syndrome, and leukemia in a randomized trial. While MOPP/ABVD and ABVD were both superior regimens to MOPP alone, ABVD was less myelotoxic than the combined regimen.
5. Dose-intense regimens have been developed with the hope of improving outcome, especially in patients with high-risk HL. The value of these regimens remains unclear.
a. BEACOPP. This aggressive 3-week cycle regimen has been compared favorably to COPP-ABVD in randomized prospective and mature studies. Higher response rates and progression-free survival are reported with dose escalation and mandatory use of growth factors, possibly with a higher risk for secondary leukemia. The effect on sterility is not fully evaluated.
b. Stanford V (
Appendix D1, Section II). Excellent results achieved with this weekly regimen in phase II studies at a single institution have not yet been confirmed in multicenter randomized trials.
c. High-dose chemotherapy followed by autologous stem cell transplantation (SCT) for patients in first remission is not generally recommended.
6. Compared effectiveness
a. A large, randomized trial conducted by a cooperative group showed that ABVD alone may be as effective as MOPP plus ABVD and more effective than MOPP alone in the management of most patients with advanced HL.
ABVD is considered the standard first-line treatment for most patients and is superior to MOPP in efficacy and toxicity profile.
b. A three-arm randomized clinical study compared COPP-ABVD with standard-dose BEACOPP and escalated-dose BEACOPP (see
Diehl et al. 2003, in “Suggested Reading”). The 5-year relapse-free rates were 69% for COPP-ABVD, 76% for standard BEACOPP, and 87% for escalated BEACOPP. The 5-year survival rate was 83% for the COPP-ABVD arm, 88% for the standard-dose BEACOPP arm, and 91% for the escalated-dose BEACOPP arm. Patients with advanced HL and adverse prognostic factors appear to derive benefit from dose escalation. BEACOPP in one of its forms can clearly be considered the treatment of choice for selected patients with high-risk HL.
7. Combined-modality treatment is becoming popular in the management of early-stage disease. The advantage of this approach is the limitation of radiation to the involved area only (and thus the reduction of the total dose), reducing long-term radiation-related complications.
a. IFRT can complement an abbreviated course of chemotherapy in patients with clinical stage I or II and nonbulky disease.
b. IFRT may be prescribed after a full course of chemotherapy to consolidate previously bulky areas of disease, especially those that respond only partially to chemotherapy. IFRT to prior sites of disease, however, may not be helpful for patients who achieve a CR with chemotherapy.
E. Treatment controversies and recommendations in classical HL (
Table 21.4)
1. Stages IA and IIA
a. Supradiaphragmatic disease. Traditionally, most patients used to undergo staging laparotomy and, if found to have pathologic stage I or II disease, would receive subtotal nodal irradiation. This approach resulted in an 80% probability of disease-free survival. Overall survival, on the other hand, may not be affected because most patients who relapse after RT can be salvaged by chemotherapy.
Excellent disease-free survival, however, has been documented after treatment with an abbreviated course of chemotherapy (two to four cycles)
followed by IFRT. ABVD regimen or the Stanford V regimen (
Appendix D1, Sections I and II) is often used. In a randomized study using four cycles of ABVD, there was no difference in outcome between groups irradiated with 2,000 or 4,000 cGy, suggesting that the dose of radiation can also be reduced.
b. Infradiaphragmatic disease. Generally, similar principles apply for early disease. Most patients could be treated with a combined-modality approach or full-course combination chemotherapy.