Histone deacetylase inhibitors (HDIs) are a promising new class of anticancer drugs that are undergoing comprehensive evaluation. They belong to the broader category of drugs that modify the epigenetic profile. Other epigenetic modifiers include the DNA methyltransferase inhibitors (5-azacytidine and decitabine) and the vitamin A analogues (retinoids). Vorinostat and romidepsin were the first HDIs to be approved for the treatment of cancer. Although not discussed here, the immunomodulatory properties of these agents are being evaluated for treatment of graft versus host disease
1 and autoimmune diseases,
2 as well as neurodegenerative conditions.
3The HDIs promote the acetylation of histones present as an octamer surrounded by DNA in the nucleosome-chromatin complex. The compaction of the complex is governed by electrostatic interactions between DNA and histones. Modification of histone at key sites, through attachment or removal of acetyl, methyl, or phosphate groups, significantly determines the accessibility of DNA to transcription complexes. The enzymes responsible for these modifications (methylases, demethylases, acetyl transferases, and deacetylases, in particular) have been implicated in tumor pathogenesis and have become targets for significant drug development efforts.
4The histone deacetylase (HDAC) enzymes appear to be the major target of the HDIs and are grouped into four different classes based on their similarity to known yeast HDACs.
5,
6 Class I HDAC isoenzymes include HDAC1, HDAC2, HDAC3, and HDAC8 and are homologous to the yeast RPD3 protein; class II HDAC isoenzymes such as HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10 are homologous to the yeast HDA1 protein; and class III HDACs are similar to the yeast SIR2 proteins. Class IV, a recent addition, contains only HDAC11. Class I and II enzymes are zinc dependent and can be inhibited by zinc chelators.
7 Class I HDACs have ubiquitous tissue expression and localize to the nucleus while class II HDACs are found in both the nucleus and cytoplasm of various tissues.
5 The class III enzymes, also known as the sirtuins, influence the longevity of model organisms,
8 but their role in cancer, tumorigenesis, and the potential therapeutic applications of modulating their activity for the treatment of cancer are poorly understood and will not be discussed here. DNA methyltransferase inhibitors are discussed in
Chapter 10; agents that alter other epigenetic modifications will not be discussed as they are still early in development.
The mechanisms underlying the ability of the majority of cancers to commandeer the epigenetic modifying pathway to manipulate gene expression are incompletely understood. HDACs exert a pro-oncogenic effect, at least in part, by keeping genes involved in differentiation, apoptosis, and cell cycle arrest in a transcriptionally quiescent state.
4,
9 Histone acetyl transferase (HAT) enzymes function in opposition to the HDACs, increasing transcriptional activity. Many cancers display molecular alterations that diminish or dampen HAT activity or shift the balance toward HDAC activity. Patients with germline mutations in CREB-binding protein (CBP), a nuclear protein with intrinsic HAT activity, have a developmental disorder known as Rubinstein-Taybi syndrome. These patients also have an increased incidence of malignant and benign neural and developmental tumors including medulloblastoma, neuroblastoma, pheochromocytoma, nasopharyngeal rhabdomyosarcoma, leiomyosarcoma, seminoma, and embryonal carcinoma.
10 Multiple alterations of the HAT proteins are found in cancers. HATs are targets of viral oncoproteins, such as HPV E6 or adenoviral E1A; chromosomal translocations involving HAT proteins are frequent in leukemias, and mutations have been identified in colorectal, breast, and ovarian cancer.
11,
12,
13,
14 Table 22-1 lists malignancies characterized by alterations in HDACs and HATs. HDIs that target these pathways have the potential to reverse mechanisms involved in oncogenesis.
Mechanism of Action
The HDIs were recognized for their anticancer properties well before they were found to interact with HDAC enzymes. Some agents were first identified based on their ability to reverse the malignant phenotype or were derived from compounds with those properties. As such, these compounds were considered differentiating agents. Erythroid differentiation was observed following exposure of cells to dimethyl sulfoxide,
15 and sodium butyrate was reported in the late 70s to induce erythroid differentiation and, subsequently, to induce histone hyperacetylation.
16,
17,
18 Multiple mechanisms have been proposed for the anticancer effects of HDIs that follow binding to and inactivation of the HDAC enzyme (
Table 22-2;
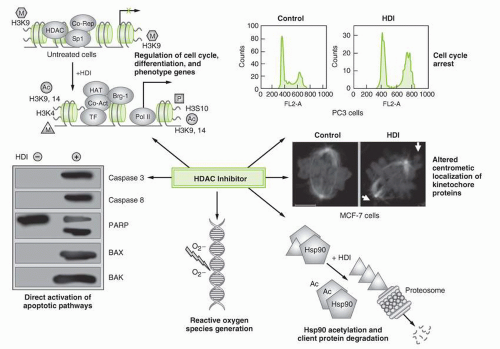
Fig. 22-1). While the HDIs affect targets in both proliferating and nonproliferating cells, they appear to preferentially affect tumor cells compared to normal cells.
19
Gene Transcription
The most widely recognized molecular effect of the HDIs has been promotion of acetylation of the amino tails of octameric histone proteins in the nucleosome core.
6,
7 HATs and HDACs have opposing
roles in the epigenetic modification that controls gene expression.
20 HDIs bind to the HDAC enzyme, thereby inhibiting deacetylation, allowing unregulated acetylation by the HATs. With an increase in the acetyl groups on histones, the positive charge on lysine is neutralized.
7 It is hypothesized that this results in a decrease in the electrostatic forces between the histone octamers and the negative DNA phosphate backbone, opening the chromatin structure and providing increased access. Transcription factors and RNA polymerases can thus more easily reach their respective regions in the now “open” chromatin. This leads to transcription of genes such as
CDKN1A, which encodes p21, and in turn leads to cycle arrest and limitation of cell growth.
7It is certain that the effects on gene transcription are physiologically much more complex, involving multiple transcription factors and downstream alterations in gene expression. The HAT proteins are part of transcriptional coactivator complexes while HDAC proteins form part of transcriptional corepressor complexes.
21 As such, the activity of transcription factors and nuclear hormone receptors is increasingly recognized to be regulated by acetylation.
22,
23 Furthermore, particular lysines are specifically acetylated or methylated as markers for activation of specific genes. This epigenetic code signals transcription of a subset of genes after exposure to the HDIs. Array studies of cancer cell lines have noted that the expression of only 2% to 5% of genes is altered upon exposure to an HDI.
24,
25,
26,
27 The number of genes induced approximately equals the number repressed. Identification of specific genes regulated and the mechanisms underlying the selective gene regulation are active areas of investigation.
Effects on Cell Cycle
Cell cycle arrest is one of the hallmarks of HDI activity. HDIs typically arrest cells in the G2/M or G1/S phase of the cell cycle.
28,
29 G1/S phase arrest has been ascribed to
CDKN1A induction (and its encoded protein p21) and cyclin D and cyclin A gene repression. These events ultimately lead to phosphorylation and decreased activity of retinoblastoma protein (pRb).
28,
30,
31 These effects are primarily mediated by induction of p21 expression.
32,
33 In addition to p21, multiple other genes involved in cell cycle progression have been identified as increased or decreased.
27,
34,
35 Cell cycle arrest may also result from activation of mitotic checkpoints following HDI-mediated acetylation of pericentromeric chromatin and impairment of kinetochore assembly.
36
Generation of Reactive Oxygen Species
Other mechanistic models include enhanced production of reactive oxygen species (ROS) by certain HDAC inhibitors as a possible mechanism for HDI cytotoxicity. Treatment of cancer cells with HDIs causes generation of ROS followed by ceramide generation, mitochondrial injury, and activation of the caspase cascade that leads to apoptosis.
37,
38 Coadministration of a free radical scavenger prevented cellular apoptosis,
37,
38 suggesting a role for ROS in cell death caused by HDIs. HDI-induced ROS generation has also been linked to up-regulation of fetal hemoglobin
39 and activation of the NFκB pathway.
40
Acetylation of Nonhistone Proteins
Acetylation of nonhistone proteins also occurs following HDAC inhibition. Indeed, this may be the principal mechanism of action in some cell types. For example, p53 is acetylated at lysines 373 and 382 following HDI exposure, causing decreased proteosomal degradation and inducing expression of p21.
41 In addition, antitumor activity may result from acetylation of c-myc, pRb, STAT-3, or NFκB, which can potentiate apoptosis.
40,
42,
43,
44,
45 This has been reviewed extensively by Marks and Xu.
46One example of nonhistone protein acetylation that has sparked particular interest results from the effects of HDIs on acetylation of Hsp90.
20 Initially demonstrated with romidepsin, acetylation of Hsp90 disrupts the interaction of the chaperone with its client proteins, including cellular molecules associated with tumor development.
47 The long list of Hsp90 client proteins (http://www.picard.ch/downloads/Hsp90interactors) includes many proteins implicated in the onset or maintenance of the malignant phenotype. Cell lines that are addicted to Hsp90 client proteins such as HER-2, the BCR-ABL fusion protein, or mutated and activated EGFR are sensitive not only to Hsp90 inhibitors but also to HDIs.
48,
49,
50 HDIs have also been shown to induce apoptosis in leukemia cell lines with mutations in the
FLT-3 gene.
51 Thus, HDIs may prove particularly active in subsets of breast or lung cancer, or leukemia, where proliferation is controlled by Hsp90 client proteins.
Inhibition of DNA Repair
HDIs disrupt DNA repair processes through acetylation or downregulation of proteins such as Ku70, Ku86, BRCA1, and RAD51. The effects of HDIs on DNA repair become apparent when combined with chemotherapy or irradiation. This could be exploited to potentiate the impact of DNA-damaging agents.
52,
53 Enhanced gamma-H2AX foci, associated with double-strand DNA repair, have been reported.
54,
55
Induction of Apoptosis
Apoptosis proceeds via one of two pathways: the extrinsic or cell death receptor-mediated pathway or the intrinsic or mitochondriamediated pathway. In the extrinsic pathway, death receptors activate caspase 8 and trigger apoptosis via a death-inducing signaling complex.
56 Several HDIs increase expression of TRAIL, DR-5, DR-4, Fas, and Fas-L.
57,
58,
59 Death receptor-mediated apoptosis is also facilitated by HDI-mediated down-regulation of c-FLIP,
57,
60 a protein associated with resistance to TRAIL therapy.
61 In the intrinsic pathway, mitochondrial release of cytochrome c leads to activation of caspase 9 that, in turn, activates caspase 3 and elicits apoptosis.
62 HDIs facilitate the intrinsic apoptosis pathway by downregulating antiapoptotic proteins such as Bcl-2, Mcl-1 and Bcl-XL and upregulating proapoptotic proteins such as Bax, Puma, and Noxa.
50,
63,
64 Up-regulation of Bim may be a key event in mediating HDI-induced apoptosis.
65In fact, the array of mechanisms reported only highlights what we do not know. Two possibilities can be considered. One is that there is a unifying mechanism of action—a single program of gene expression that leads to cell death depending upon the proclivity to apoptosis. The limited activity of the HDIs in solid tumors, as opposed to hematological malignancies, would support this model. The diverse gene expression profiles observed in different cell types after HDI exposure argue somewhat against this, although the possibility exists that the totality of altered gene expression is intolerable to the cell.
The other possibility is that mechanism depends upon specific cellular context. A T-cell lymphoma may find gene induction of death receptors intolerable and undergo rapid apoptosis. A cell with amplification of ErbB2 or mutation or addiction to the epidermal growth factor pathway may be more sensitive to the loss of Hsp90 chaperone function and client protein degradation than a cell without.
47,
50 A myeloma cell filled with paraproteins may be more sensitive to the loss of HDAC6-mediated transport to the aggresome.
66 The impact of varying mechanism of action could offer a spectrum of possibilities for the use of HDIs in the clinic.
One question that regularly arises is whether there are differences among the HDIs currently in development, which predominantly inhibit class I enzymes. Evidence to date suggests that inhibition of the class I enzymes is the predominate mechanism of action. HDI antitumor activity is duplicated when either HDAC1 or HDAC2 is down-regulated by siRNA; and abrogated with overexpression of these genes.
67,
68,
69 However, to the extent that the various HDIs display differing IC
50s across the family of isoenzymes, different efficacy or toxicity profiles could result. For example, HDAC6 is thought to participate in the trafficking of ubiquitinated, degraded proteins to the aggresome.
70 Differing affinities for HDAC6 could lead to differential effects on this cell protective function.
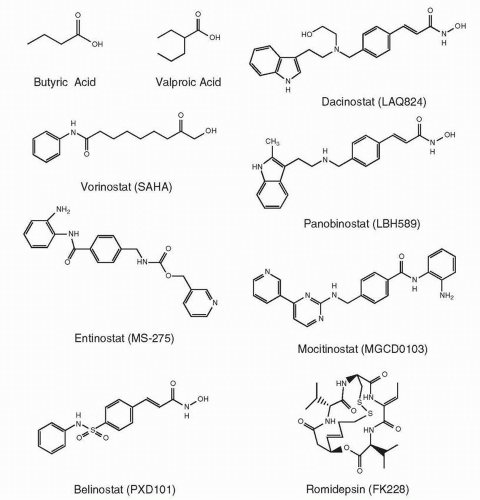
Chemical Structures and Classification of HDIs in Clinical Use
The HDI family includes a number of structurally unrelated compounds grouped together by their common mechanism of action and classified according to their chemical structures. HDIs generally contain a zinc-binding domain, a capping group, and a straight
chain linker connecting the two.
46 A partial list of HDIs currently in clinical trials is provided in
Table 22-3, and structures of some of these agents are provided in
Figure 22-2.
The first class of agents recognized as HDAC inhibitors and tested clinically were the short chain and aromatic fatty acids (sodium butyrate,
71 phenylbutyrate,
72 and valproic acid
73). Sodium butyrate has been approved for the treatment of urea cycle disorders and valproic acid for seizure disorders. Valproic acid is also used in the treatment of manic depression and in migraine prophylaxis. Pivaloyloxymethyl butyrate (AN-9), a prodrug of butyric acid, is currently in clinical trials.
Vorinostat or suberoylanilide hydroxamic acid (SAHA), N1-hydroxy-N8-phenyl-octanediamide, belongs to the hydroxamic acid class of HDIs that includes PCI-24781; givinostat (ITF2357), {6-[(diethylamino)methyl]naphthalen-2-yl}methyl [4-(hydroxycar-bamoyl)phenyl]carbamate; panobinostat (LBH589), (E)-
N-hydroxy-3-[4-[[2-(2-methyl-1H-indol-3-yl)ethylamino]methyl]phenyl] prop-2-enamide; dacinostat (LAQ824), 3-[4-[
N-(2-Hydroxyethyl)-
N-[2-(1H-indol-3-yl)ethyl]aminomethyl]phenyl]-2(E)-propenohydroxamic acid; and belinostat (PXD101),
N-hydroxy-3-(phenylsulphamoylphenyl) acrylamide. The path to the discovery of vorinostat began with the observation that dimethylsulfoxide was able to induce synthesis of hemoglobin and differentiation of murine erythroleukemia cells.
15 Synthesis and screening of related polar compounds ultimately led to the selection of vorinostat as the lead compound.
74 It was the structural similarity to trichostatin A that provided the first clue that it was an inhibitor of HDACs.
74,
75 The key features of vorinostat are shown in
Table 22-4.
Romidepsin, (1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-bis(1-methyletheyl)-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8.7.6] tricos-16-ene-3,6,9,19,22-pentone, also known as FR901228, FK228, and depsipeptide, is a bicyclic peptide. First isolated from the fermentation broth of
Chromobacterium violaceum76,
77 based on its ability to induce differentiation of H-ras-transformed NIH 3T3 cells, it was later shown to potently inhibit HDACs.
76 Subsequent studies revealed it to be a substrate of P-glycoprotein
78, and clinical development at the National Cancer Institute was initiated based on its unique structure, mechanism, and favorable toxicity profile. The disulfide bond of romidepsin must be reduced inside the cell to yield the drug’s active form.
79 The key features of romidepsin are shown in
Table 22-5.
The benzamide class of HDIs, including entinostat (MS-275), (pyridylmethyl-N-{4-[(2-aminophenyl)-carbamoyl]-benzyl}-carbamate); tacedinaline (CI-994), 4-(acetylamino)-N-(2-aminophenyl)benzamide; and mocetinostat (MGCD0103), (N-(2-aminophenyl)-4-((4-(pyridin-3-yl)pyrimidin-2-ylamino)methyl) benzamide), are highly potent HDIs now undergoing clinical evaluation.
Pharmacokinetics
Pharmacokinetic studies of HDIs have been useful in guiding route of administration and schedule. They have identified clinical differences among the HDIs, with implications for toxicity and efficacy.
An important challenge in using the short chain fatty acids is the need to achieve millimolar concentrations to achieve induction of histone acetylation, gene expression, or apoptosis (reviewed in Gore and Carducci
80). Extensive pharmacokinetic data are available for valproic acid. It has nearly 100% bioavailability, reaching peak plasma concentrations between 50 and 100 mg/L (0.35 to 0.7 mM) between 1 and 4 hours. It has a volume of distribution of 0.2 L/kg with about 90% protein binding with CSF concentration reaching about 10% of plasma levels. The half-life in plasma ranges from 7 to 15 hours. It is metabolized primarily in the liver, undergoing beta and omega oxidation via P450 mixed oxidases. Metabolism may be enhanced with concomitant use of agents that induce cytochrome P450 enzymes and may be prolonged with hepatic insufficiency. Valproic acid is predominantly eliminated by excretion, with about 7% enterohepatic recirculation. Clinical trials of valproic acid as an HDI have tried alternate schedules and formulations, with inconclusive efficacy. Maximum plasma concentrations up to 300 mg/L at the maximum tolerated dose (MTD) have been reported.
81,
82While initially tested by the intravenous route, oral administration has become the predominate route in the clinical development of vorinostat.
83,
84 Based on initial studies, the estimated oral bioavailability of vorinostat is 43%; oral administration with a high fat meal resulted in increased absorption.
84 While many dose levels have been studied, the dose used for the FDA registration trial in patients with cutaneous T-cell lymphoma (CTCL) was 400 mg PO qd. At steady state, the area under the concentration versus time curve (AUC) is 6.0 ± 2.0 μM × hour,
Cmax 1.2 ± 0.53 μM, and median
Tmax 4 hours. The plasma half-life is estimated to be
between 1.5 and 2.0 hours.
84 Vorinostat is 71% bound to human plasma proteins over the range of concentrations of 0.5 to 50 μg/mL. Vorinostat undergoes biochemical transformation in the liver through phase I hydrolysis and beta-oxidation and phase II conjugation detoxification by glucuronidation to form metabolites such as vorinostat
O-glucuronide and 4-anilino-4-oxobutanoic acid.
85 CYP450 enzyme involvement appears not to be significant. Though the mean steady-state concentrations of these metabolites are much higher compared to the parent drug, they are pharmacologically inactive.
Other compounds of the hydroxamic analogue class are in advanced stages of clinical evaluation. Belinostat has also been tested in both IV and oral formulations. With IV administration, three-compartment pharmacokinetics is proposed. Pharmacokinetics are linear for
Cmax and AUC independent of dose.
86 Nonhuman primate studies show minimal, less than 1%, penetration into cerebrospinal fluid.
87 Animal studies suggest oral bioavailability at 30%. Elimination half-life is independent of dose and ranges from 0.3 to 1.3 hours. The major elimination route is through the bile, and only 0.2% to 2.0% of parent drug was excreted in urine.
86