Fig. 21.1
(a) Cutaneous venulocapillary malformation (port-wine stain). (b) Leptomeningeal venulocapillary malformation, Sturge-Weber Syndrome
Venous Malformations
Clinical
Terms used in the past to refer to lesions now recognized as venous malformations (VM) include cavernous hemangioma and venous hemangioma. Use of these outdated terms as diagnostic labels for lesions that are biologically consistent with VM is confusing to clinicians (and inaccurate) and should be abandoned. It is of interest that so-called cavernous hemangiomas or cavernomas of the brain, terms still firmly entrenched among neurosurgeons, are localized collections of massively dilated capillaries or venules, rather than veins.
Venous malformations are typically singular lesions, either localized or segmental, with no associated abnormalities, although some occur as part of complex syndromes. Superficial lesions appear blue in color and enlarge under conditions that increase venous pressure (e.g., dependency or exertion). Extensive lesions may be complicated by chronic, low-grade consumptive coagulopathy, and phleboliths are common.
Multiple venous malformations occur as a familial mucocutaneous disorder and also in the poorly understood dysmorphic syndrome known as blue rubber bleb nevus syndrome, first described in 1958 by Bean [13]. Blue rubber bleb nevus syndrome comprises an association between multiple venous malformations of the skin and the gastrointestinal tract, complicated by gastrointestinal bleeding. Venous malformations have also been described in patients with Turner’s syndrome [14]. Multiple mucocutaneous venous malformations inherited as an autosomal dominant trait have been linked to a locus (VMCM1) on chromosome 9p21 and are associated with activating missense mutations in the endothelial cell-specific TIE-2 tyrosine kinase gene [15, 16, 17]. Recent studies have also identified somatic TIE-2 mutations in a large percentage of sporadic venous malformations [17, 18]. Multiple VM also occur without recognized etiology in Maffucci syndrome, a rare congenital disorder also characterized by dyschondroplasia, multiple enchondromas, and spindle cell hemangiomas. Similarly, multiple gastrocutaneous VM seen in blue rubber bleb nevus syndrome are associated with gastrointestinal bleeding and anemia; some cases are sporadic, and others autosomal dominant, all without known etiology.
Microscopic Features
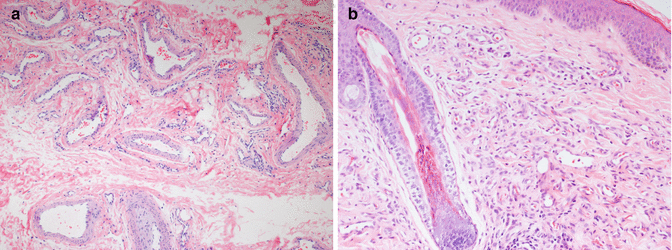
Venous malformations are characterized by abnormal collections of veins that are superficial or deep, diffuse or localized, and solitary or multiple. They have flat, mitotically inactive endothelial cells (Fig. 21.2a). The venous nature of these malformations is implied by the presence of a variable amount of well-differentiated smooth muscle in the vessel walls (usually scant relative to luminal diameter), the absence of an internal elastic membrane, and erythrocyte-filled lumina. Vessels of capillary or venular proportions may also be present within the lesion. Component veins vary with regard to luminal size and wall thickness both between and within individual lesions. Those excised from some patients, particularly with Klippel-Trénaunay syndrome, may show extreme disorganization of component smooth muscle fibers. Lining endothelial cells are positive for CD31, vWF, and CD34 and are negative for GLUT1 and other IH-associated markers [5].
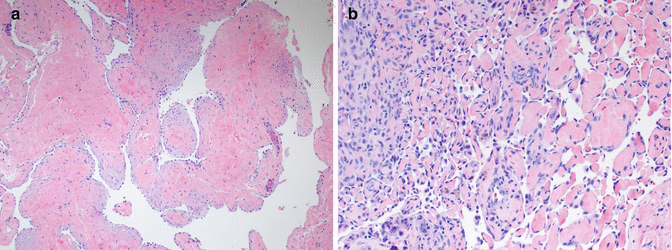
Fig. 21.2
(a) Venous malformation. (b) Venous malformation, intravascular papillary endothelial hyperplasia
Luminal thrombi in various stages of organization are common in venous malformations, reflective of stasis in these low-flow lesions. Recanalizing thrombi may demonstrate intravascular papillary endothelial hyperplasia (Fig. 21.2b). Although the latter may appear concerning for possible malignancy, this histological pattern represents an exuberant response of endothelial cells to organizing thrombus. It was first described by Masson in 1923 in hemorrhoidal veins and has been referred to as Masson’s tumor or Masson’s pseudoangiosarcoma. Early stages of this organizing process show growth of endothelial cells into fibrinous thrombus material, which divides it into papillary fronds lined by a single layer of plump endothelial cells without significant cytologic atypia. In later stages, the fibrin cores of the papillae become collagenized and hyalinized, and the endothelial lining becomes attenuated. Fusion of the lesional papillae may form an anastomosing meshwork of vessels separated by connective tissue stroma reminiscent of angiosarcoma; the pleomorphism, necrosis, and relatively high mitotic rate of angiosarcoma are lacking. This type of lesion can also present as a mass within an apparently normal vein, most commonly in adults, and rarely as an organizing extravascular hematoma. Foci of intravascular papillary endothelial hyperplasia are common in VM and help distinguish these low-flow lesions from high-flow arteriovenous malformations.
Glomuvenous Malformations
Lesions characterized by presence of benign glomus cells have been subclassified historically into categories such as diffuse type, solitary type, multiple type, solid type, adult type, and pediatric type. Current evidence supports division of these lesions into two major categories: (1) the glomus tumor proper, a cellular neoplasm that tends to be well circumscribed, solitary, and subungual, and (2) the so-called glomangioma, a frequently multifocal lesion, more properly termed glomuvenous malformation, that presents in infants or children and histologically resembles a venous malformation in which lesional vessels are surrounded by layers of glomus cells. The following discussion is restricted to this second category, which accounts for 10–20 % of all glomus cell lesions.
Clinical
Glomuvenous malformations (GVMs) are superficial lesions becoming evident in childhood or adolescence and generally covering a large area of skin and/or subcutis. They appear either as multiple, widely distributed to confluent, soft, red-to-blue nodules, or as pink to deep blue multifocal plaques. Although clinically resembling venous malformations, GVMs differ in a number of ways. They tend to be more nodular or cobblestone-like in appearance, appear bluer and less compressible, and do not swell with exercise or dependency [19].
GVM is less painful than the glomus tumor proper but may be tender to palpation, and attacks of pain may occur during menstruation and pregnancy. All reported cases have behaved in a benign fashion. Due to their more expansive, multifocal nature, GVM is less amenable to surgery than common adult-type glomus tumors and may recur locally following subtotal resections and may progress locally [20]. Sclerotherapy is less effective for GVMs than for venous malformations [20, 21]. Most GVM is sporadic; some rare familial cases have demonstrated an autosomal dominant pattern of inheritance [22, 23]. Based on linkage disequilibrium studies with these families, a locus for glomangiomas (termed VMGLOM) has been mapped to chromosome 1p21-p22 and codes for a protein of still uncertain function termed glomulin [24–26]. Sporadic glomangiomas likely result from somatic mutations at this locus.
Microscopic Features
GVM consists of dilated, thin-walled veins in the dermis and subcutis, often distributed as separate nodules. They are histologically similar to those comprising venous malformations but are surrounded by one or more layers of cuboidal glomus cells (Fig. 21.3). The glomus cell component can be quite variable from region to region and vessel to vessel, making adequate sampling important. Like venous malformations without glomus cells, many GVMs contain organizing thrombi or phleboliths.
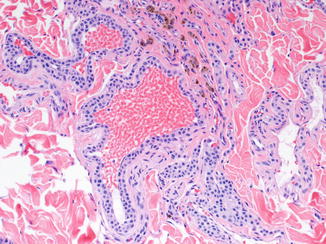
Fig. 21.3
Glomuvenous malformation
Arteriovenous Malformations
Clinical
Like all vascular malformations, arteriovenous malformations (AVMs) are the result of errors in vascular morphogenesis and are not neoplastic in origin. Use of the outdated term “arteriovenous hemangioma” is thus inappropriate. AVM is usually evident at birth and associated with an often clinically significant degree of arteriovenous shunting. Those with superficial extension may produce a palpable thrill or pulsation. Clinically significant hemorrhage and local tissue ischemia due to arterial steal are relatively common. Deep lesions may not become apparent until later in childhood or even adolescence or adulthood if shunting is low grade. AVMs tend to progress over time as collateral arterial flow is recruited into the low-resistance vascular bed. For the same reason, they often recur with a vengeance if incompletely excised or inadequately embolized. Unlike AV fistulas, which are often acquired lesions and are characterized by one or a few large AV shunts, AVMs are more complex developmental anomalies with a myriad, perhaps millions, of small abnormal AV connections that bypass a normally controlled, high-resistance vascular bed.
The vast majority of AVMs are sporadically occurring single lesions of unknown etiology. AV shunts are seen less commonly in familial disorders such as hereditary hemorrhagic telangiectasia (HHT) and the capillary malformation-arteriovenous malformation disorder (CMAVM). CMAVM, characterized by multiple atypical cutaneous capillary stains and increased incidence of arteriovenous malformations, is caused by inactivating mutations in RASA1 [27]. HHT, also known as Osler-Rendu-Weber syndrome, is an autosomal dominant disorder characterized by multisystemic angiodysplasia leading to frequent epistaxis, telangiectasias, GI bleeding, and arteriovenous shunts in the liver, brain, and lung. It has been linked to mutations in two genes and has therefore been designated HHT type 1 (HHT1; gene, endoglin; chromosome 9q34.1) and HHT type 2 (HHT2; gene, ALK–1, chromosome 12q11-q14) [28, 29]. HHT in association with juvenile polyposis has been linked to mutations in SMAD4 [30].
Microscopic Features
The histological appearance of AVMs often varies considerably from one area to another, and the actual arteriovenous shunts are difficult to find without extensive sectioning or special techniques. Most histological sections show beds of arterioles, capillaries, and venules within a densely fibrous or fibromyxomatous background, intermixed with numerous larger caliber arteries and thick-walled veins (Fig. 21.4a). The arteries are often tortuous, and the veins typically show adventitial fibrosis and irregular intimal fibrosis. There is no evidence of thrombosis or intravascular papillary endothelial hyperplasia, consistent with the abnormally high venous flow. Involved skin often contains a prominent “pseudoangiosarcomatous” proliferation of small vessels, creating a ragged, cellular appearance that lacks the delicate lobularity of IH (Fig. 21.4b). Isolated foci of mitotically active small vessel proliferation reminiscent of a vascular tumor such as infantile hemangioma or pyogenic granuloma can also be seen in more deeply seated regions of many AVM, admixed with the large vessel component. This proliferative component may dominate the histological picture in deep intramuscular AVMs, particularly those involving the tongue, potentially leading to a misdiagnosis of hemangioma. These unusually cellular AVMs, like the more typical forms of AVM, are negative for GLUT1 and other infantile hemangioma-associated antigens [5, 31]. Clinical and radiological correlation with histology is essential in the diagnosis of AVM.