Heterogeneity in Cancer: The “Cancer Stem Cell” Hypothesis
13.1 INTRODUCTION: CAN WE IDENTIFY THE CANCER CELLS THAT KILL PATIENTS?
Every patient’s cancer is different. Cancers arising from the same organ have different histology and metastatic proclivity, are more or less aggressive, and have different responses to therapy. There are many interdependent mechanisms and dimensions of heterogeneity that account for this variability between cancers. There is also heterogeneity within individual cancers, between stromal cells with a normal genome and mutated malignant cells, between differently mutated malignant clones, between epigenetically different subpopulations within clonal populations, and between cells within different microenvironments within the tumor (Fig. 13–1; see Chap. 12, Sec. 12.2). Recognition of this heterogeneity gives rise to the intriguing possibility that a subset of cancer cells are resistant to treatment, may cause primary tumor recurrence or seed distant metastasis, and may be identifiable a priori. A surgeon’s concern in ensuring complete resection of primary tumors (“negative margins”) where more invasive cancer cells may reside, the prognostic relevance of circulating tumor cells (see Chap. 10, Sec. 10.3.4), and the resistance of disseminated micrometastases to adjuvant chemotherapy are phenomena that might be explained by “special” cells within a cancer, so-called cancer stem cells, cells that must be targeted to achieve long-term remission or cure. The cancer stem cell (CSC) hypothesis states that only a minority of cancer cells has the potential to (a) self-renew, (b) proliferate indefinitely, and (c) differentiate to give rise to more differentiated tumor cells (Reya et al, 2001). This chapter addresses the competing models that attempt to account for this epigenetic heterogeneity, led by the CSC hypothesis, but first describes the stromal and genetic heterogeneity of human cancers.
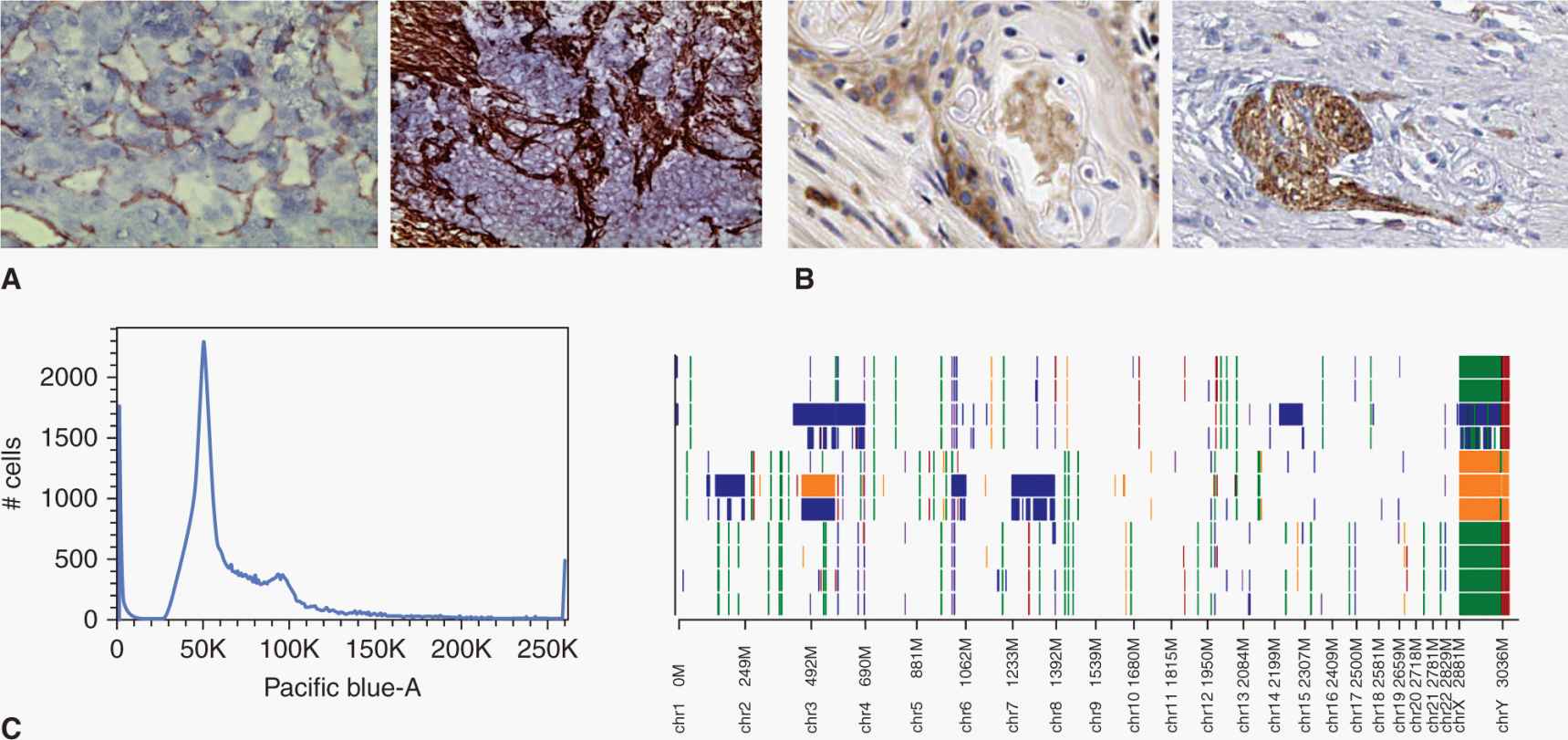
FIGURE 13–1 Cancers are heterogeneous. This heterogeneity must be represented and considered in many interdependent dimensions. For example, (A) cancers show stromal heterogeneity as many cells within cancers are nonmalignant (left, CD31+ endothelial cells (brown) in renal carcinoma; right, collagen-positive fibroblasts (brown) in ovarian carcinoma), (B) epigenetic heterogeneity, where malignant cells have different phenotypes that can be associated with different functional behaviors (left, CD44+ cells (brown) in head and neck carcinoma; right, N-cadherin–positive cells (brown) in colorectal carcinoma), and (C) genetic heterogeneity, where different clonal populations within cancer have different genomic mutations (left, distribution of cells with different DNA content analyzed by flow cytometry (Pacific blue-A staining is proportional to DNA content); right, DNA microarray showing gains (blue, green) and losses (orange, red) of DNA across different chromosomes (X-axis), which shows the “copy-number variation” against the normal human genome (background white).
13.2 HETEROGENEITY IN CANCER
13.2.1 Stromal Heterogeneity in Cancer
Every seed needs appropriate soil to germinate and grow, and all cancers have a stromal component that supports, and often protects, malignant cells. Cancer cells recruit, manipulate, and nurture this microenvironment to establish and maintain their nutrition and survival. Stromal cells include endothelial cells that line tumor blood vessels and lymphatics; pericytes to support mature endothelium in larger vessels; cancer-associated fibroblasts that both provide physical scaffolding and interact with and influence malignant cells; and tumor-associated macrophages, lymphocytes, and other leucocytes. Cancer-associated fibroblasts produce an extracellular matrix (ECM) consisting of collagen and various other molecules (see Chap. 10, Sec. 10.2.1). The stromal component of a tumor can be sparse, for example, in melanoma or large cell lymphomas where the malignant population dominates, or profuse, such as in pancreatic adenocarcinoma, which is typically dominated by a thick fibrous stroma. At the extreme is Hodgkin lymphoma, where rare malignant Reed-Sternberg cells sit isolated in a sea of reactive inflammatory cells (Fig. 13–2). This variability in the tumor microenvironment influences the effectiveness of cancer treatments and may itself be a legitimate target of cancer treatment (see Chap. 19, Sec. 19.3). It also raises the prospect that CSCs may be treatable by influencing their interaction with the stromal niche (see Sec. 13.5.3).
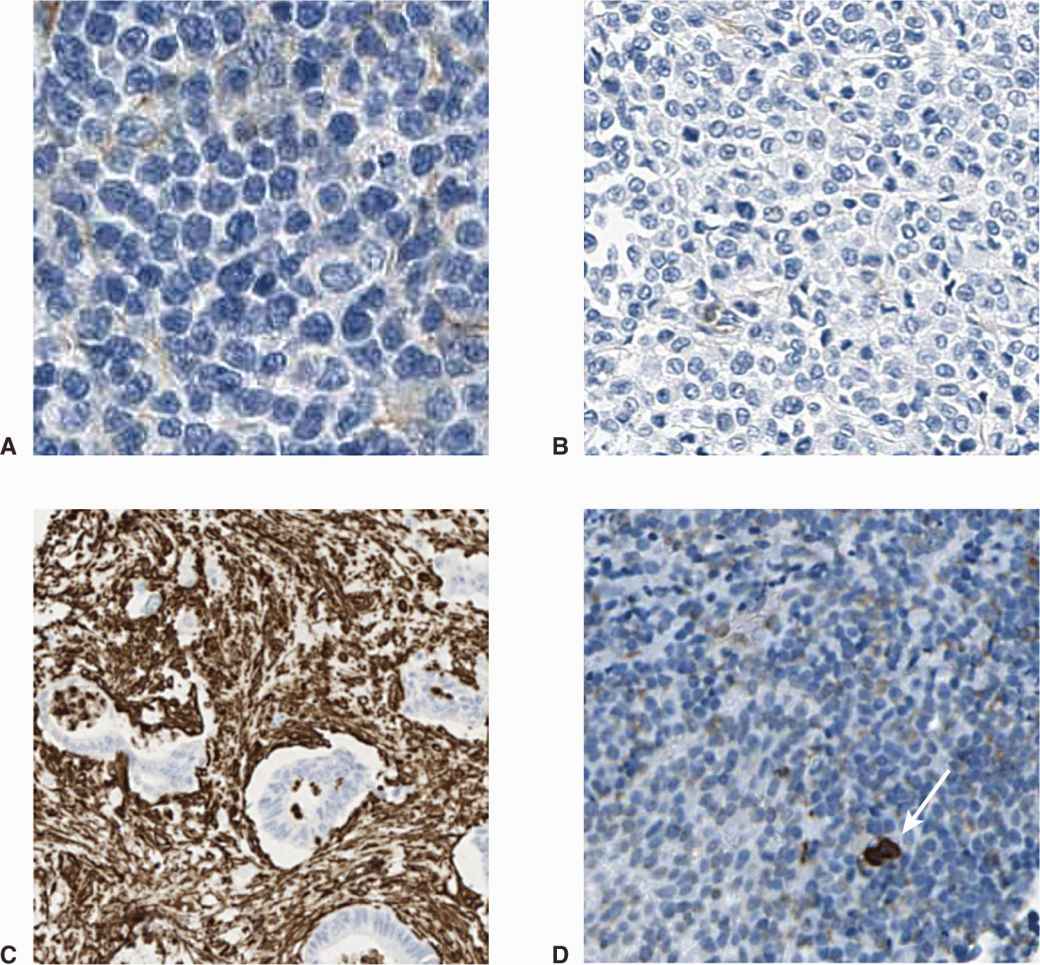
FIGURE 13–2 Different cancers have different stromal microenvironment. For example, in large cell lymphoma (A) and melanoma (B) the cancer exists as sheets of tumor cells, with few stromal cells present. In contrast, some cancers have a dense infiltrate of genomically normal fibroblasts or leucocytes (C), pancreatic adenocarcinoma with stroma stained brown) and Hodgkin lymphoma (D), with rare malignant Reed-Sternberg cell stained for the cell surface marker CD15 [arrow]).
13.2.2 Genetic Heterogeneity in Cancer
Most cancers contain multiple identifiable mutations in their cells. Genetic heterogeneity between different patients’ cancers is believed to be a major determinant of variable histology and of clinical behavior. For example, patients with microsatellite instability in colorectal carcinoma may have a better prognosis, but conversely, their tumors may be more unresponsive to chemotherapy. This may occur because microsatellite unstable tumors tend to have more mutations than other colorectal cancers (Kucherlapati et al, 2012), and this greater genetic complexity may allow for a more adaptable genetic evolution when exposed to the selection pressure of chemotherapy. The different driver mutations in individual patients’ cancers are becoming important in selecting treatment—examples include the selective use of hormonal agents and human epidermal growth receptor (HER)-2–targeted therapies in subgroups of women with breast cancer, and the use of BRAF and c-KIT inhibitors in different subsets of melanoma (see Chap. 7, Sec. 7.5).
Individual cancers are thought to arise from the sequential acquisition of multiple mutations, each, in turn, conferring a survival advantage to a clonal population of cells, an evolutionary process first proposed by Nowell (1976) (see Chap. 10, Sec. 10.1.1). This process has perhaps best been documented in colorectal cancer where the transition from adenoma to dysplastic adenoma to carcinoma to invasive and metastatic carcinoma has been shown to parallel the acquisition of mutations in genes such as APC, K-RAS, PI3KCA, and TP53 (Jones et al, 2008). This progression is almost certainly a stochastic process as many different genetic clones can be identified in the premalignant lesions associated with various cancers, for example, in melanoma, renal cell carcinoma, colorectal carcinoma, and pancreatic carcinoma (Campbell et al, 2010). The evolution of intratumoral cancer clones may not be sequential; for example, multiple genetically distinct leukemia-initiating cell subclones have been demonstrated in children with acute lymphoblastic leukemia. These diverse clones have differing abilities to generate xenografts, and if the dominant subclone is capable of growth in the mouse microenvironment, this portends a poor clinical outcome (Notta et al, 2011). With the advent of faster, cheaper, and more detailed DNA sequencing (see Chap. 2, Sec. 2.2.10), rarer clonal populations can be identified in many cancers (Fig. 13–3). Rapid sequencing of thousands of single cancer cells has identified various clonal patterns in breast carcinoma, including very small clonal populations with highly aberrant genomes (Navin and Hicks, 2011). There is also evidence that as cancers progress to an advanced stage that a dominant clone usually predominates within the primary tumor and in metastases, presumably as a result of a process of genetic selection. In men with metastatic hormone-refractory prostate cancer, for example, examination of the cancer genome performed by single-nucleotide polymorphisms in DNA microarrays of the in situ primary and multiple bone metastases showed that most cancers were monoclonal across all sites (Liu et al, 2009). Whether cancer progression, treatment resistance, and clinical failure are caused by properties inherent in the dominant clone or by outgrowth and rapid selection of rarer subclones, is likely to be variable but of critical importance to each cancer patient’s management.
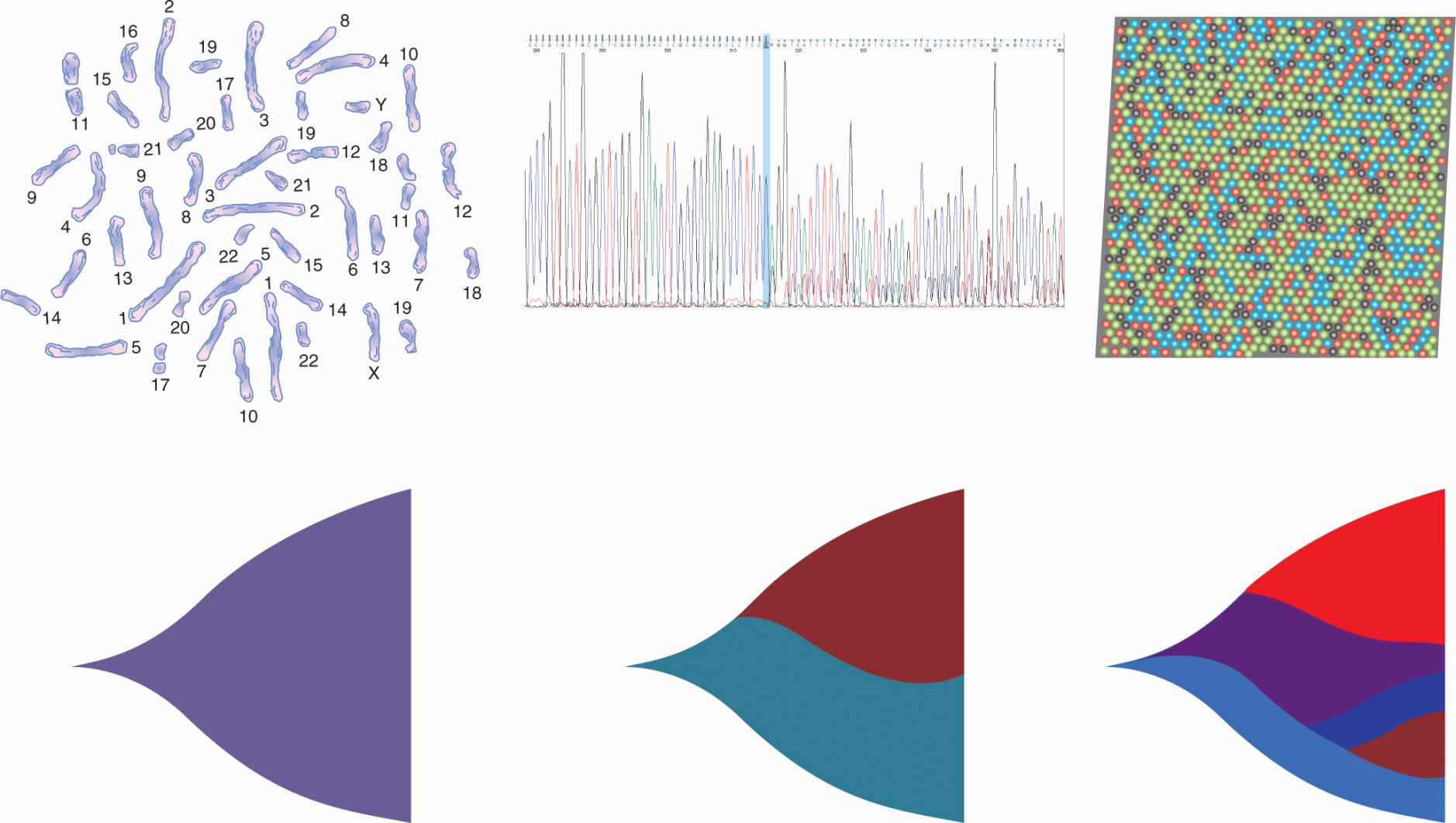
FIGURE 13–3 Increasingly detailed genomic data from successive generations of DNA sequencing technology reveal the clonal structure of cancer cell populations. Early techniques, such as karyotyping, allowed recognition of large chromosomal changes, while single base pair sequencing showed the primary sequence of these and other mutations. Second- and third-generation technologies, such as pyrosequencing and bridge amplification (“next-gen sequencing” where every nucleotide base-pair is individually digitally counted by changes in red, blue or green fluorescence), allow “deep” sequencing to detect rare clones (represented in different colors here to show their outgrowth and evolution from previous clones) within tumor cell populations, and even parallel sequencing of single cancer cell clones.
13.2.3 Intratumoral Epigenetic Heterogeneity
Although mature cancers often contain a dominant clone of malignant cells, the properties of individual malignant cells in this clone may be variable. These properties are governed by epigenetic changes that influence the expression of genes within the common genome (see Chap. 2, Sec. 2.3) and by microenvironmental influences (see Chap. 12, Sec. 12.2).
Well-established, clinically apparent cancers represent a relatively late stage in the development of a malignancy. Each gram of cancer may contain half a billion cancer cells, and may shed large numbers of cancer cells into the blood circulation each day. Evidence that only a small proportion of these mobilized tumor cells leads to clinical metastases (see Chap. 10, Sec. 10.3.3) begs the question whether all cancer cells have equivalent potential to form metastases given the appropriate microenvironment (a stochastic process), or whether different cancer cells have intrinsically different self-renewal and differentiation potentials (ie, whether there is an intratumoral hierarchy (Fig. 13–4). Many theories have been proposed to account for this epigenetic heterogeneity but 2 are dominant: (a) stochastic production of tumor cells with different properties, often linked with evidence of epithelial-to-mesenchymal transition (EMT) and (b) the hierarchical CSC hypothesis. The main focus of this chapter is on the CSC hypothesis, but it first discusses EMT.
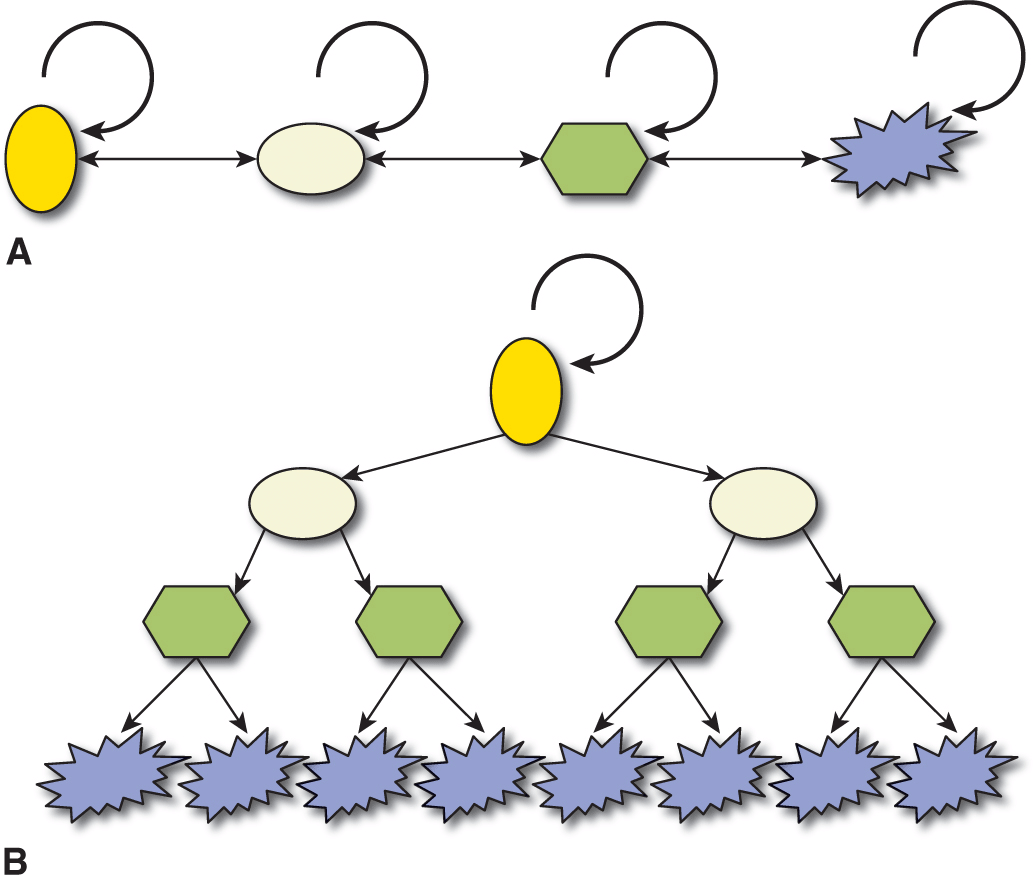
FIGURE 13–4 Two mutually exclusive theories can be proposed to account for epigenetic tumor heterogeneity. A) Every cancer cell might have the potential to self-renew (circular arrow) to maintain the primary tumor or form a new metastasis, if it receives appropriate microenvironmental signals. Tumor maintenance or dissemination is therefore a random or stochastic process, and variation between different behaviors may involve transition to and from epithelial to mesenchymal phenotypes. B) Conversely, cancers may be organized hierarchically in a disordered parody of normal stem cell hierarchies, where only a subpopulation of CSCs has the potential to self-renew, proliferate extensively, and differentiate to form the bulk of the tumor consisting of non–stem cancer cells with a terminally differentiated phenotype. By definition, non–stem cells cannot dedifferentiate to CSC.
13.3 EPITHELIAL-TO-MESENCHYMAL TRANSITION
EMT is a process that is described in a variety of biological scenarios to account for the ability of cells to transdifferentiate from one morphology and function to another, specifically to transform from a polarized epithelial phenotype to a motile, mesenchymal cell phenotype (Kalluri and Weinberg, 2009) (Fig. 13–5; see also Chap. 10, Sec. 10.5.6). The reverse of this process is termed mesenchymal-to-epithelial transition (MET), and both processes occur in embryogenesis and wound healing and are proposed to occur in cancer.
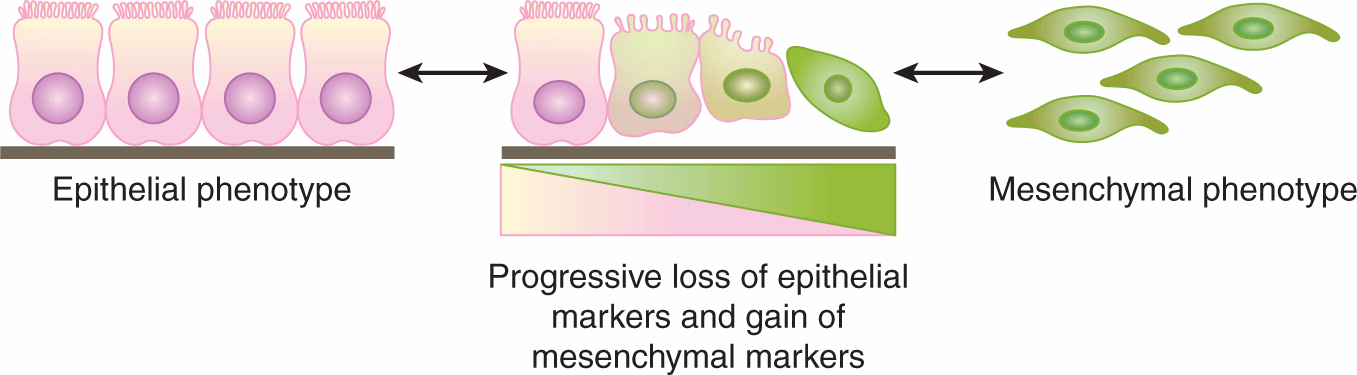
FIGURE 13–5 The EMT describes the process of transformation of cells with a polarized, epithelial phenotype to cells with a motile, mesenchymal phenotype. This process is thought to occur during normal development and wound healing, and there is increasing evidence that similar processes occur in cancer. (Adapted from Kalluri and Weinberg, 2009.)
EMT has been studied in many in vitro and model systems and involves molecular events that give rise to morphological and functional alterations of cell behavior. Through the influence of extracellular signals from molecules such as transforming growth factor (TGF)-β, Wnt, and platelet-derived growth factor (PDGF) (see Chap. 8, Sec. 8.4), cells with a polarized epithelial phenotype upregulate the expression of proteins and molecules that drive the cell to reorganize its cytoskeleton (eg, desmin), disconnect from underlying ECM (eg, metalloproteinases), and migrate through the local environment or more distally via vascular channels (eg, vimentin; see Chap. 10, Sec. 10.5).
EMT is thought to occur in at least 3 situations: embryogenesis, wound healing, and malignancy. EMT might occur in cancer because mutated cancer cells may be more easily influenced by microenvironmental signals, as receptors for these molecules may be overexpressed, and downstream signaling pathways may be constitutively activated by the acquisition of further mutations. Many aspects of EMT have been translated from development and wound healing to cancer biology, but there is criticism of the EMT concept as it applies to malignancy, because much of the evidence is based on mouse models, in vitro experiments, and correlative pathological studies (Tarin, 2005).
Evidence for EMT in cancer includes the observation that cultured human epithelial cancer cells can express markers of EMT, such as loss of E-cadherin expression, and upregulation of expression of N-cadherin, smooth muscle actin, vimentin, TWIST, SNAIL, and S100A4. Cancer cells expressing these markers can be found at the “leading edge” of patients’ tumors during histopathology, and are thus hypothesized to account for the population of cancer cells that can be shed into the circulation and become metastatic (Brabletz et al, 2001; see also Chap. 10, Sec. 10.5), and this phenomenon has been directly observed in xenograft models (Stoletov et al, 2010). There is also clinical evidence for the relevance of EMT in cancer. For example, markers of EMT are associated with poor prognosis in gastric carcinoma (Ryu et al, 2012), renal cell carcinoma (Mikami et al, 2011), breast carcinoma (Nes et al, 2012), and non–small cell lung carcinoma (Hung et al, 2009). Expression of EMT markers in cancer cells is also associated with resistance to treatment, including cytotoxic chemotherapy, hormonal agents, and drugs targeting growth factor signaling pathways such as epidermal growth factor (EGF) and HER2. It is unclear if EMT can be targeted to improve cancer management, but several studies suggest that targeting microRNAs (see Chap. 2, Sec. 2.4.3) associated with EMT (such as the miR-200 family) may assist in differentiating mesenchymal-phenotype cancer cells to epithelial-phenotype cells, which may then be more susceptible to chemotherapy (Wang, Li et al, 2010).
13.4 CANCER STEM CELLS
13.4.1 The Cancer Stem Cell Hypothesis
The CSC model posits that a hierarchy exists within each individual patient’s cancer, such that only a subset of malignant cells has the potential to self-renew, proliferate extensively, and differentiate to recapitulate the primary tumor’s morphological and antigenic heterogeneity (Ailles and Weissman, 2007). Conversely cancer “non–stem cells” may retain some limited proliferative potential but do not to have the ability to propagate the primary tumor or seed distant metastases. The CSC model essentially represents tumors as caricatures of renewing tissues, such as the bone marrow, intestine, and skin, where it is well established that a limited number of stem cells has the capacity to self-renew and to produce progeny that differentiate and carry out the normal function of that tissue (see Chap. 12, Sec. 12.1.3). When applied to cancer, the CSC hypothesis has important implications: If only a subset of cells within a tumor is able to repopulate or seed fresh cancers, then these cells are the true targets for therapy, and if current therapies fail to eliminate these cells, then even apparent complete clinical responses will be noncurative. Instead of a cancer being compared to an infection such as an abscess, where all bacteria must be eliminated to cure an infection, cancer may be viewed rather like the organizational hierarchy of a beehive, with a queen bee, workers, and drones; only survival of the queen bee will allow reestablishment of the colony. Not every CSC is fated to form a new metastasis (Shackleton et al, 2009). CSC remain subject to the influence of the stromal microenvironment, for example to the degree of vascularization or local hypoxia, to the ambient pH and composition of the ECM. For example, although the murine melanoma cell line B16 is an aggressive cancer model where almost every cell is functionally a CSC, only 1 in 100 cells can survive to form metastases in an intravenous metastasis model (Luzzi et al, 1998).
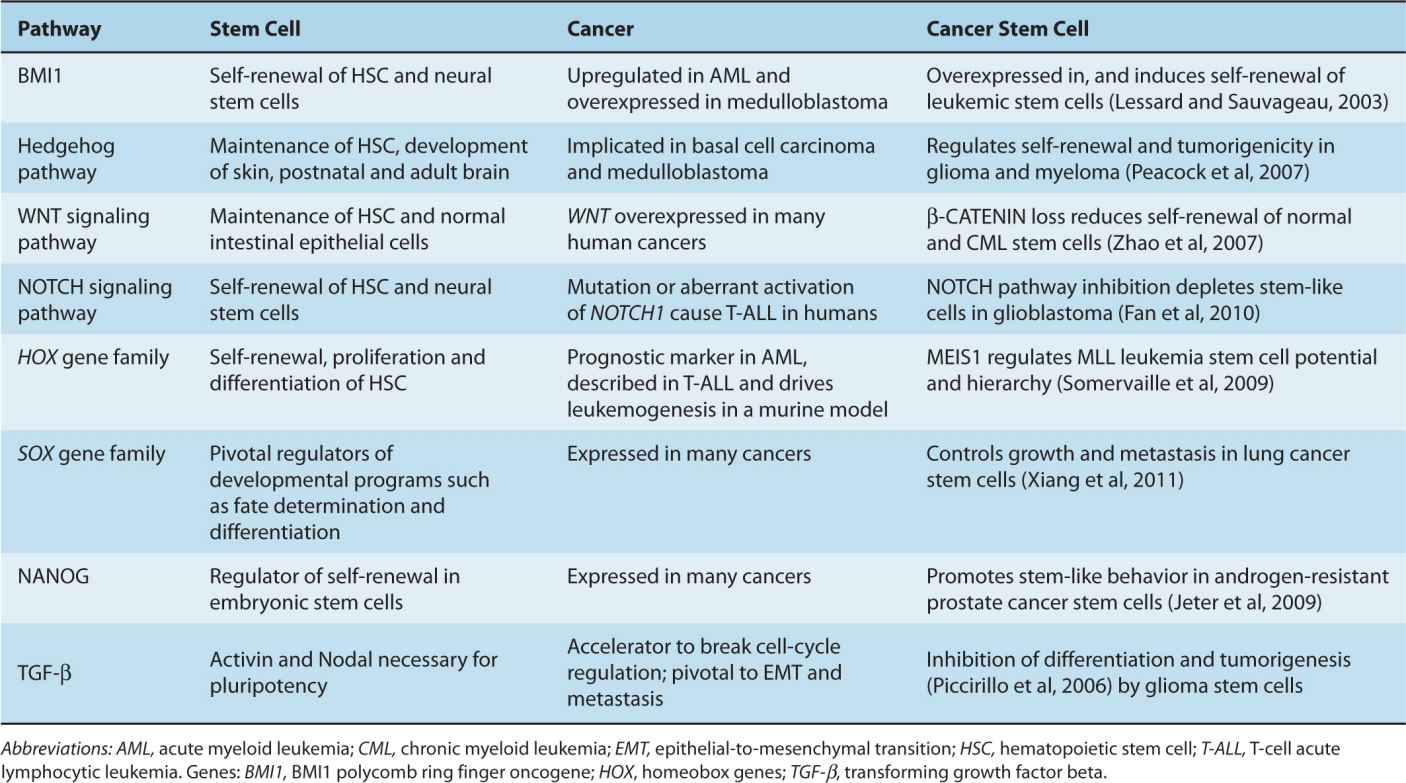
There is controversy regarding the CSC hypothesis, in part because of misconceptions about the model. First, the CSC hypothesis does not imply that somatic stem cells in the corresponding normal tissue are necessarily the cells-of-origin for cancer. Somatic stem and progenitor cells are likely to be the cell-of-origin in some (perhaps most) cancers (eg, medulloblastoma, germ cell tumors), but some terminally differentiated cells, which can be much longer-lived than previously assumed (Rawlins and Hogan, 2008), may have sufficient time to acquire oncogenic mutations in a stepwise fashion, allowing them to evade apoptosis and senescence, proliferate independently of microenvironmental signals, and reactivate their self-renewal potential. The variation in histology, differentiation, and phenotype in cancers arising from a single tissue may be partly a result of different cells of origin. Second, the expression in cancer of genes and pathways that are expressed and active in embryonic stem cells and normal somatic stem cells, such as OCT4 (octamer-binding transcription factor 4, also known as POU5F1), NANOG, BMI1 (BMI1 polycomb ring finger oncogene), and the Hedgehog, NOTCH, and WNT signaling pathways (see Chap. 8, Sec. 8.4) is not proof of the existence of “cancer stem cells.” Their reexpression in cancer most likely reflects the parsimonious use of molecular programs useful for self-renewal, asymmetric cell division, dormancy, migration/metastasis, proliferation, and differentiation, which are properties of both somatic stem cells and cancer cells (Table 13–1). Expression of these “stemness” genes has been invoked in other models of epi-genetic heterogeneity in cancer, such as BMI1 in EMT (Yang et al, 2010), and their presence does not imply a hierarchical organization in cancer.
TABLE 13–1 Shared signaling pathways in stem cells, cancer, and cancer stem cells.
The CSC hypothesis and other models based on epigenetic mechanisms of heterogeneity are not mutually exclusive. As much of the experimental data to support the existence of CSCs is based on samples taken from “late” clinical human cancers, it is difficult to determine whether the CSC hypothesis can account for events within early carcinogenesis. Stromal, epigenetic, and genetic mechanisms of tumor heterogeneity are likely to be involved in every clinical cancer. Unraveling the interconnections between these different mechanisms is an important challenge in cancer research.
13.4.2 Historical Perspective of the CSC Hypothesis
Paget’s recognition that metastases “seed” into organs that provide a fertile “soil” provided the conceptual framework that tumor cells must interact with their microenvironment, and that some tumor cells may be more fit than others for this process (Paget, 1889). Pierce and his colleagues demonstrated that teratocarcinomas and mouse squamous cell carcinomas contained highly tumorigenic cells that could differentiate into morphologically differentiated cell types that were unable to form tumors when transplanted in mice (Pierce and Wallace, 1971). He described cancers as a “caricature” of normal tissue renewal, whereby tumor stem cells divide and differentiate giving rise to terminal postmitotic differentiated cells (Pierce and Speers, 1988). Till and McCulloch provided the first experimental evidence of normal tissue stem cells (Till and McCulloch, 1961), when they injected lethally irradiated mice with donor mouse bone marrow to determine the minimum number of cells required to repopulate the marrow of the recipient mouse. Colonies of cells from all 3 blood lineages were seen to form in the spleen and bone marrow of recipient mice. When these single-cell-derived colonies were subsequently transplanted into secondary recipient mice, some clones could again give rise to blood cells from multiple lineages. A frankly unethical experiment from the 1950s provided the first clinical evidence that tumor formation is limited to a subpopulation of human cancer cells. Cancer cells from patients with ovarian, cervical, and uterine cancer were cultured in vitro following palliative surgery and reinjected into the same patient subcutaneously (Brunschwig et al, 1965). Even when a million cancer cells were implanted, only approximately 50% of these injections developed into a palpable nodule. Hamburger and Salmon (1977) demonstrated in clonogenic assays that only a fraction of cells from freshly excised human tumors could form colonies in tissue culture (including myeloma, lymphoma, neuroblastoma, ovarian carcinoma, chronic lymphocytic leukemia, small cell lung cancer, and melanoma). This result led the authors to speculate that these rare clonogenic cells had been tumor stem cells in vivo, although their results might have been caused by an inadequate environment to support growth in tissue culture. Other scientists demonstrated that only a fraction of mouse lymphoma cells were capable of forming colonies in an irradiated mouse (Bruce and Van Der Gaag, 1963). Hewitt (1979) also demonstrated that large numbers of cells were needed to transplant spontaneously arising murine tumors into syngeneic mice, again implying that the putative CSC was rare.
13.4.3 Methods Used to Study Cancer Stem Cells
CSCs are defined both by their ability to generate tumors upon serial transplantation in immune-deficient animals (ie, high reproductive capacity) and by their ability to recapitulate the genetic and phenotypic heterogeneity of the original tumor (Fig. 13–6). The proposed CSC subpopulation should demonstrate potent tumor initiation by regenerating the tumor when a limited number of cancer cells are injected. Evidence for CSC self-renewal is obtained by observing xenograft formation after serially transplanting reisolated CSC into secondary and tertiary recipient mice. At each stage, larger doses of marker-negative, cancer “non–stem cells” (eg, CD133– or CD44–; see below) should fail to engraft in mice. The second criterion is that the injected CSC must differentiate and recapitulate the phenotype of the tumor from which they were derived, although multilineage differentiation as seen in normal somatic stem cells is not a requirement for transplanted cells to be labeled as CSC. Based on the above properties, CSC are defined functionally as tumor-initiating cells (TICs), xenograft-initiating cells, or cancer-initiating cells.
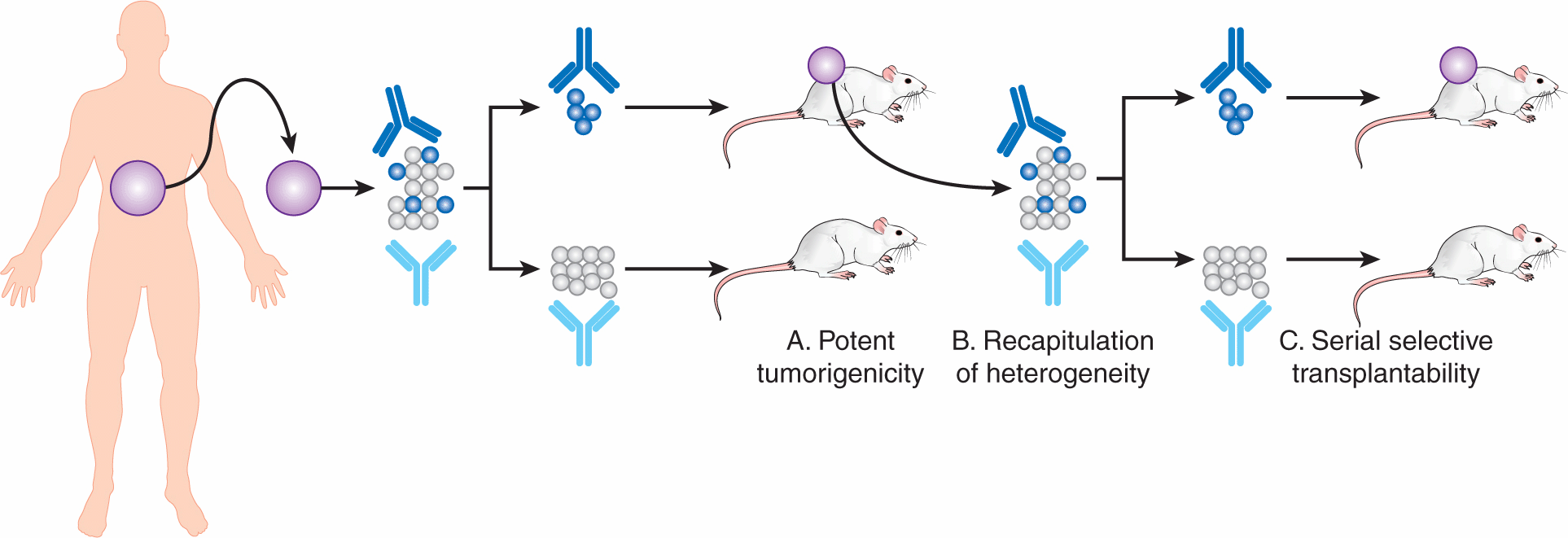
FIGURE 13–6 The CSC hypothesis is tested experimentally using the serial xenotransplantation assay. CSCs are functionally defined by their ability to (A) potently initiate xenograft tumors in immunocompromised mice, which (B) recapitulate the heterogeneity and phenotype of the original patient tumor, and upon reisolation from the xenograft (C) retain this selective tumorigenicity in serial xenograft experiments.
CSCs must be identifiable a priori, and a variety of markers can be used to separate cancer and normal cells, many of which are cell-surface proteins. CSC markers have been discovered by trial and error, but have frequently been translated from normal stem cell biology to the cancer setting. For example CD133 (prominin-1 [PROM1]) is a marker for neural stem cells and has been applied to neuroectodermal tumors such as glioblastoma (Lenkiewicz et al, 2009). Cancer cells are usually obtained from solid human tumors donated by patients immediately after their surgical removal. Tumors are usually mechanically disaggregated, then digested to a single cell suspension by use of enzymes such as collagenase, hyaluronidase and DNase (Ailles et al, 2009). The resulting single-cell suspension can be examined by flow cytometry, cultured briefly in vitro, or injected into immunocompromised mice (Fig. 13–7). Flow cytometry uses fluorescently labeled antibodies to measure semiquantitatively proteins and antigens on intact cells. Viable cells can also be sorted (fluorescence-activated cell sorting [FACS]) into different subpopulations (Alexander et al, 2009). For example stromal cells in tumors can be obtained by sorting CD45+ hematopoietic cells or CD31+ endothelial cells. From the remaining cancer cells one may then identify a population of cells suspected of being CSCs, for example, by selecting CD133+ cells (Fig. 13–8). Magnetic cell separation uses antibodies conjugated to tiny paramagnetic beads, which allow cells to be physically separated in a high magnetic field (Palmon et al, 2012). Cell function and behavior can also be used to identify CSC. For example, the “side-population” method takes advantage of the ability of normal stem cells and CSC to efflux water -soluble fluorescent dyes such as Hoechst 33342. More dormant, slow-cycling, infrequently dividing cells can be identified by staining a cell population with lipophilic fluorescent dyes, such as PKH26, then selecting fluorescent cells after a period of time in culture; CSC are thought to be less likely to divide and are therefore brighter. Finally, CSC can be isolated using a fluorescent dye that is only liberated inside cancer cells expressing aldehyde dehydrogenases (ALDHs), enzymes that are preferentially expressed in normal and CSCs.
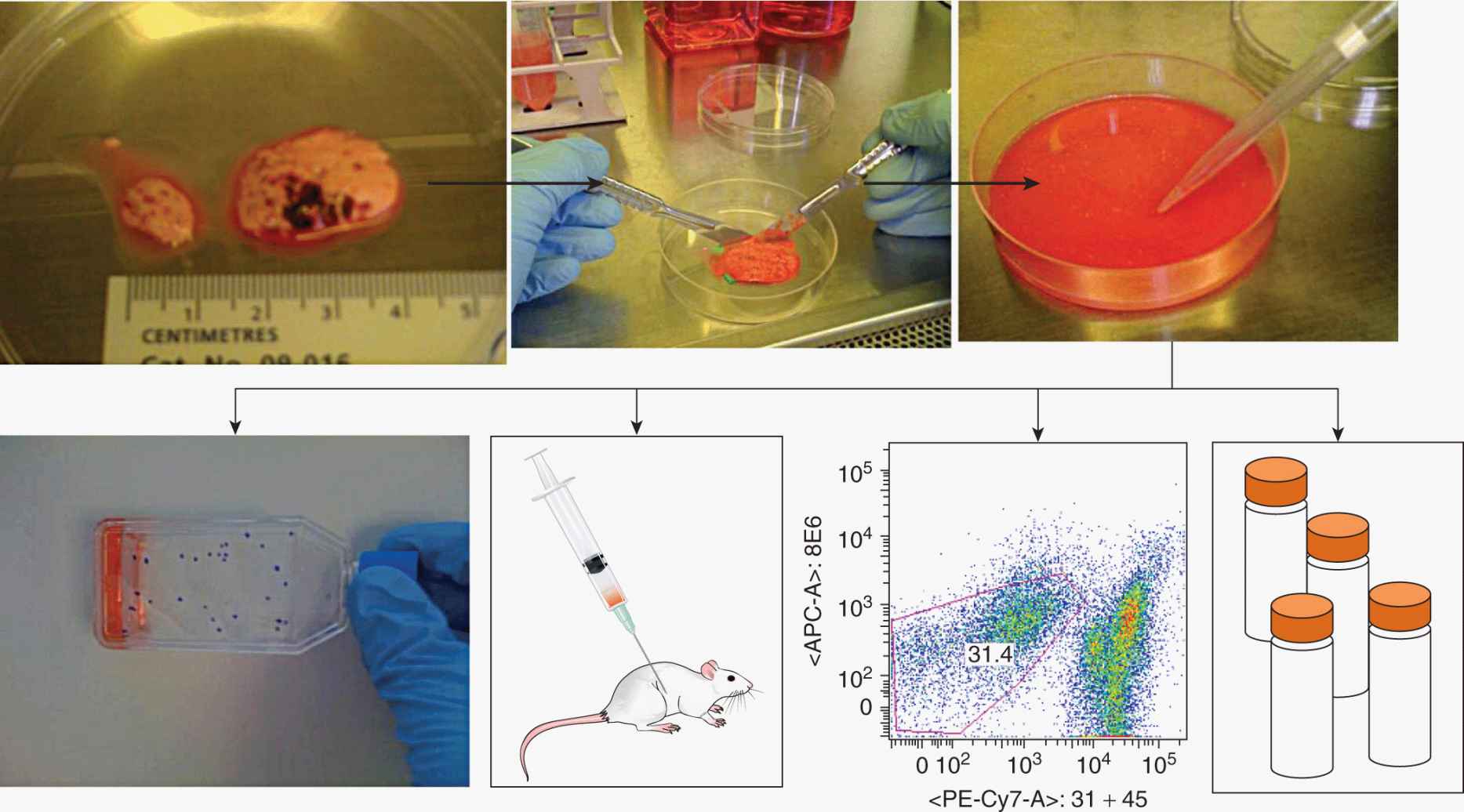
FIGURE 13–7 Methods to study the CSC hypothesis in solid tumors. Cancer samples are mechanically dissociated and then enzymatically digested and filtered to generate a suspension of single cells. This single cell suspension can be used for a variety of experiments, for example, to establish new cancer cell lines, to measure the tumor-forming ability of these cancer cells by injection into immunocompromised mice, or analysis by flow cytometry (in this example ~30% of cells are cancer cells, while ~70% of cells express hematopoietic, endothelial or fibroblast markers). Finally, cells can be saved for later use by cryogenic freezing.
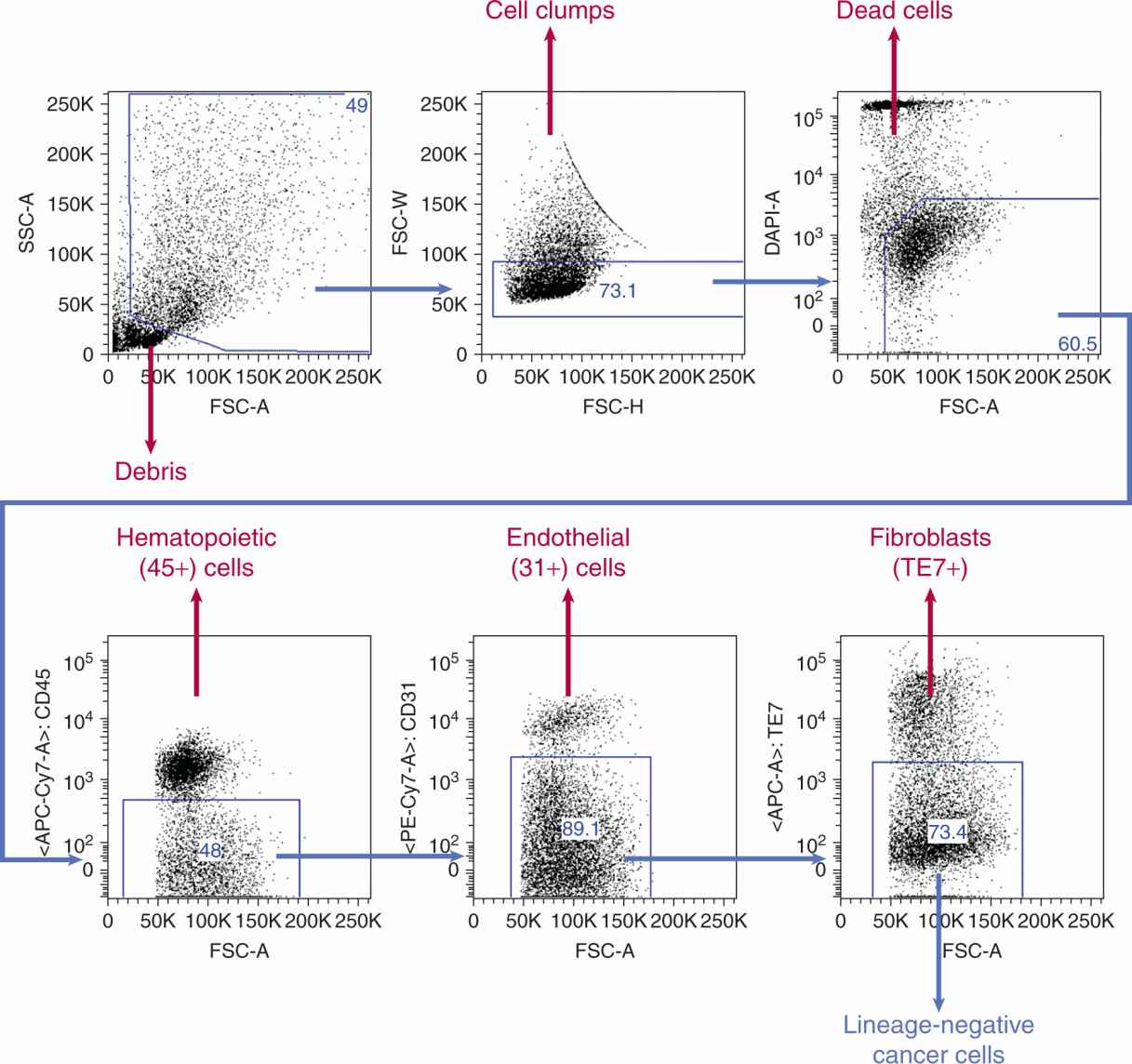
FIGURE 13–8 Fluorescence-activated cell sorting (FACS) allows the selection of normal stromal and malignant cancer cells from solid tumors and leukemias. After separating off debris (ie, cells that have clumped together, or cells that are dead), antibodies to cell surface markers, such as CD45 and CD31, can be used to separate hematopoietic and endothelial cells, respectively. Each blue “gate” is a subset of cells from the previous plot. For example, just over half of cells in this sample (bottom left panel) are white blood cells, and of the remaining 48% of cells, ~10% are endothelial cells. Remaining cells that do not express a lineage marker, so-called lineage-negative cells are the remaining cancer cells, from which proposed CSC populations can be selected by markers such as CD133 and CD44. FSC and SSC represent forward scatter and side scatter, optical properties of the cells that help discriminate their size and complexity. DAPI, APC-Cy7, APC, and PE-Cy7 are all fluorescent dyes used to identify different cell populations. Percent of cells in the boxed areas are indicated.
Once cancer cells have been processed, characterized, and segregated, they can then be used for in vivo and in vitro experiments. These experiments can evaluate their ability to form xenografts in mice by limiting dilution assay, and can assess the smallest number of cells injected that are capable of forming a tumor.
Early experiments in stem cell biology used lethally irradiated mice (Till and McCulloch, 1961) to investigate repopulation of normal hematopoietic stem cells of the same inbred strain of mice, but investigation of human cancer cell biology requires immunocompromised mice. Athymic nude mice have been employed as recipients of xenografts since the 1960s, but these mice have residual antitumor immunity because of natural killer (NK) cell activity, and better engraftment is observed using the severe combined immunodeficiency (SCID) mouse that lacks T and B lymphocytes. Improved engraftment was seen when the SCID mouse was crossed with the nonobese diabetic (NOD) mouse model, yielding NOD/SCID mice. These immunocompromised mouse models have been refined further, using mice that have mutation in the Rag2 or inter-leukin-2 receptor γ chain (IL2Rγ-/-) genes or similar mice crossed with the NOD/SCID mouse forming the NOD/SCID/IL2Rγ-/- or NSG mouse, which lack NK-cell activity as well as B- and T-cell activity.
Injections into NOD/SCID or NSG mice are often performed in conjunction with Matrigel, a heterogeneous mixture of basement membrane proteins secreted by mouse sarcoma cells, which enhances tumor transplantability. Whether this enhancement is a result of tumor cell aggregation within a gel or a result of growth factors embedded in the matrix remains unclear, but it does not appear to be caused by collagen or laminin components of the matrix alone. Finally, mouse xeno-graft experiments can be further refined using various forms of “humanization,” either via incorporation of human tissue, such as fetal bone or adult skin, or the addition of human growth factors.
Although the functional definition of a CSC is assessed via in vivo tumor-forming experiments, surrogate in vitro assays may allow for rapid screening of drug activity and interrogation of signaling pathways in CSCs. There are criticisms of these cell-culture models, particularly as the microenvironment for cancer cells growing in a culture dish is very different to that within the primary tumor. For example, glioblastoma cells grown in bovine serum acquire in vitro mutations that transform them into cell lines with genotypes and phenotypes that do not represent the genotype and phenotype of the patients’ tumor (Lee et al, 2006). This property may contribute to the often quite large differences between the phenotypes and genotypes of cell lines derived from human cancers and the ex vivo tumors themselves (van Staveren et al, 2009).
Improvements to these models are evolving. For example, human tumor cells cultured de novo as “tumor spheres” in defined serum-free media (Brewer et al, 1993) can generate cell lines with morphological heterogeneity that express markers of a primitive stem-like phenotype and form xeno-grafts that are invasive and maintain the genotype of the original tumor. Cell lines grown in defined media have been employed to demonstrate the efficacy of targeting glioblastoma stem cells by various stem-cell-signaling pathways, but most of these in vitro models are not well validated in representing properties of CSC (Pollard et al, 2009). Once tumors or cell lines have formed, their genotype, phenotype, and differentiation status must be studied to see that it remains constant with each subsequent passage, and that it represents an accurate reproduction of the original patient’s tumor. Flow cytometry, immunohistochemistry, messenger RNA (mRNA) expression by polymerase chain reaction (PCR) or gene expression microarray and genotype by karyotype or copy-number variation microarray are all methods in common use (see Chap. 2).
Finally, techniques to study the epigenetic regulation of CSCs are improving rapidly and should provide further mechanistic data on the validity of the CSC hypothesis (and EMT). Whole-genome methylation arrays, whole-genome histone modification arrays, microRNA, and noncoding RNA arrays are becoming increasingly comprehensive (see Chap. 2, Sec. 2.6.2), and suitable for small samples that are typically available when studying CSCs obtained directly from a patient’s tumors.
13.4.4 Cancer Stem Cells in Hematological Malignancies
The renaissance of the CSC hypothesis followed the isolation of leukemia-initiating cells in acute myeloid leukemia (AML) (Lapidot et al, 1994; Fig. 13–9). Leukemic cells from the bone marrow and peripheral blood of patients with AML could be engrafted in SCID mice supplemented with human stem cell factor (SCF) and a human granulocyte-macrophage colony-stimulating factor (GM-CSF) interleukin-3 (IL-3) fusion protein. Some mice developed a morphologically and histochemically identical disease to the original patient. Large doses of AML cells were required for engraftment. In vitro clonogenic assays showed similar rare frequencies (0.3% to 0.9%) of AML colony-forming units (CFUs) in primary patient samples or from AML cells extracted in bone marrow aspirates from transplanted mice, suggesting that the potential to engraft leukemia into SCID mice remained a rare property. The authors then examined the cell-surface phenotype of AML cells using flow cytometry, and discovered that AML cells expressing a phenotype similar to normal human hematopoietic stem cells (CD34++/CD38–) were much more likely to engraft in SCID mice than CD34+/CD38+ or CD34– cells. In more immunocompromised NOD/SCID mice, which have far fewer NK lymphocytes, AML cells could engraft at much lower doses (10- to 20-fold). Using established statistical methods, the SCID leukemia-initiating cell (SL-IC) assay was shown to be reproducible, although there was a wide variation in the SL-IC frequency from different patients, suggesting underlying differences between their leukemias. Subsets of AML cells were again isolated by flow cytometry and injected into NOD/SCID mice. CD34+ cells were able to engraft whereas CD34– cells did not, and the CD34++/CD38– AML subset were highly enriched for SL-IC. As few as 5000 CD34++/CD38– cells could engraft a NOD/SCID mouse, whereas 5 × 105 CD34+/CD38+ could not. AML cells isolated from mice injected with CD34++/CD38– leukemia stem cells showed a similar pattern of cell-surface markers (immunophenotype) and similar morphology compared to the original cancer, implying in vivo differentiation of engrafted SL-IC. Finally, evidence of in vivo self-renewal of SL-IC was obtained, showing that CD34++/CD38– cells could be isolated from primary xenografted animals and transplanted into secondary recipients with similar efficiencies of engraftment and recapitulation of ex vivo phenotype (Lapidot et al, 1994).
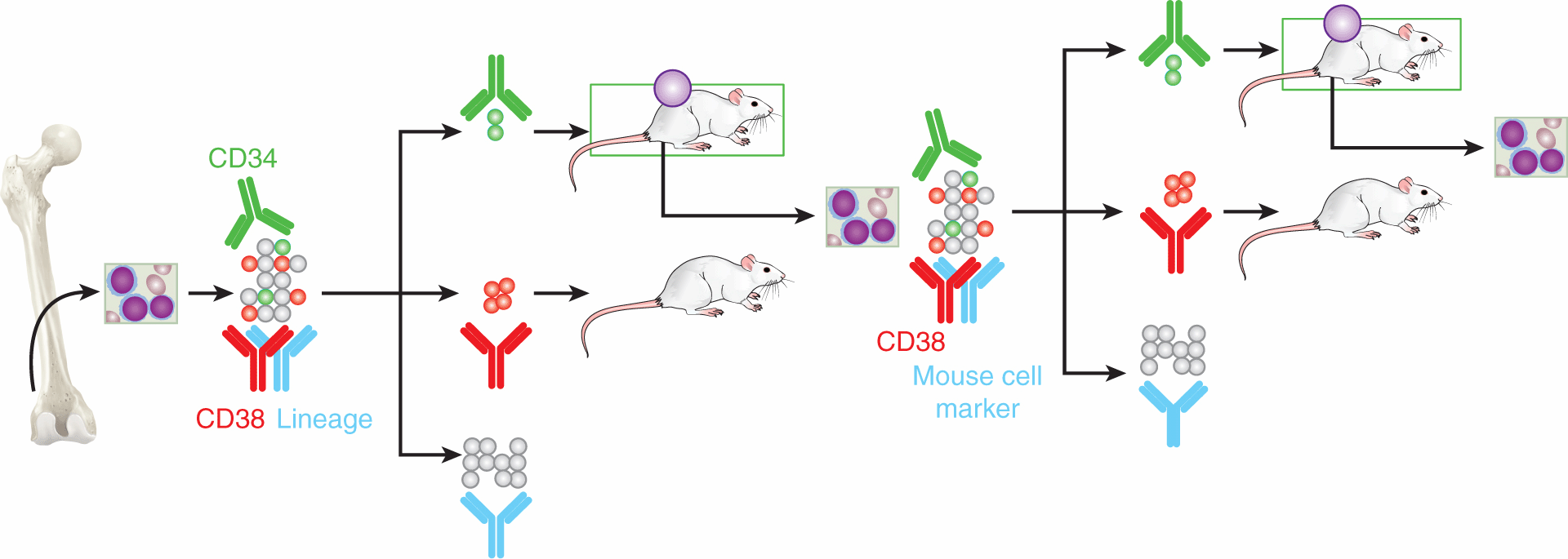
FIGURE 13–9 CSC in hematological cancers. Using cell-surface markers established from normal hematopoietic stem cell biology, leukemic stem cells were discovered in a series of elegant experiments; these leukemic cells were negative for the progenitor cell marker CD38, but expressed the normal stem cell marker CD34. To accurately measure the “stem” and “non-stem” cancer cells, other antibodies (in blue) are used to identify and segregate non-cancerous cells (eg, lymphocytes and macrophages) from the human sample (the “lineage” cells) and mouse cells from the xenografted leukemia sample. Mice that engraft the leukemia are represented here with a purple circle. Cells could be serially passaged through several generations of mice, and maintained the same phenotype and morphology as the leukemia from the original patient.
Bonnet and Dick (1997) extended these findings by demonstrating that similar to the hierarchy within normal hematopoietic stem cells, that there is a “hierarchy within the hierarchy” of human AML leukemic stem cells (LSCs). Donor AML populations were transfected with a lentivirus encoding green fluorescent protein (GFP). The lentivirus stably incorporates into the genome of individual cells at a random location, and Southern blotting can be used to track this insertion site, thus allowing tracking of each clone in each mouse. This genetically engineered protein was used as a fluorescent marker to separate human AML cells from unmarked mouse cells, and also provided a means of tracking clonal progeny of an individual AML cell through serial xenografts in recipient mice. From serial transplantation of samples from multiple patients into mice, it became apparent that there were several different populations of LSCs, which the authors termed short-term, long-term, and quiescent long-term SL-ICs. In some cases, quiescent SL-IC did not contribute to leukemogenesis until transplanted into a third serial mouse, and after 6 months ex vivo. Clonal tracking demonstrated that long-term SL-IC gave rise to short-term SL-IC, and that quiescent long-term SL-IC gave rise to both short-term and long-term SL-IC, demonstrating a “hierarchy within the hierarchy” and conclusive evidence of CSC within AML. The similarity of this hierarchy to normal hematopoiesis suggests that the cell-of-origin for AML may be a normal hematopoietic stem cell (Hope et al, 2004).
Chronic myeloid leukemia (CML) provides a compelling example of the CSC model in clinical practice. Since the discovery of the oncogenic BCR-ABL translocation (see Chap. 7, Sec. 7.5.1), and the development of small-molecule tyrosine kinase inhibitors such as imatinib (see Chap. 7, Sec 7.7 and Chap. 17, Sec. 17.3.1), survival in this disease has increased substantially. However, despite the rapid disappearance of malignant cells from the blood of patients treated with these agents, there is evidence that CML stem cells remain dormant within the bone marrow, and that a fraction of these CML stem cells are resistant to BCR-ABL inhibition and can undergo further clonal evolution to give rise to blast crisis (Chu et al, 2011).
CSCs have proven more challenging to study in other hematological malignancies, such as lymphomas and myeloma. This may be related to the broad heterogeneity in genetic, histochemical, and clinical subtypes in lymphoma, and perhaps also because of the fastidiousness of these cancers for their microenvironment. For example, addition of a humanized microenvironment improves engraftment and growth of human myeloma in immunocompromised mouse models, perhaps by providing an appropriate niche or bone marrow stromal cells that can secrete supportive human cytokines (Tassone et al, 2005).
13.4.5 Cancer Stem Cells in Solid Tumors
CSCs have been identified in many solid cancers, including squamous cell carcinoma of the head and neck (Prince et al, 2007), colorectal cancer (Dalerba et al, 2007; O’Brien et al, 2007; Ricci-Vitiani et al, 2007), pancreatic carcinoma (Hermann et al, 2007; Li et al, 2007), hepatocellular carcinoma (Ma et al, 2007; Yang et al, 2008), ovarian carcinoma (Zhang et al, 2008; Stewart et al, 2011), and prostate carcinoma (Collins et al, 2005; Patrawala et al, 2006), as well as cancers of the breast, brain, and melanoma that are discussed in detail below (Fig. 13–10).
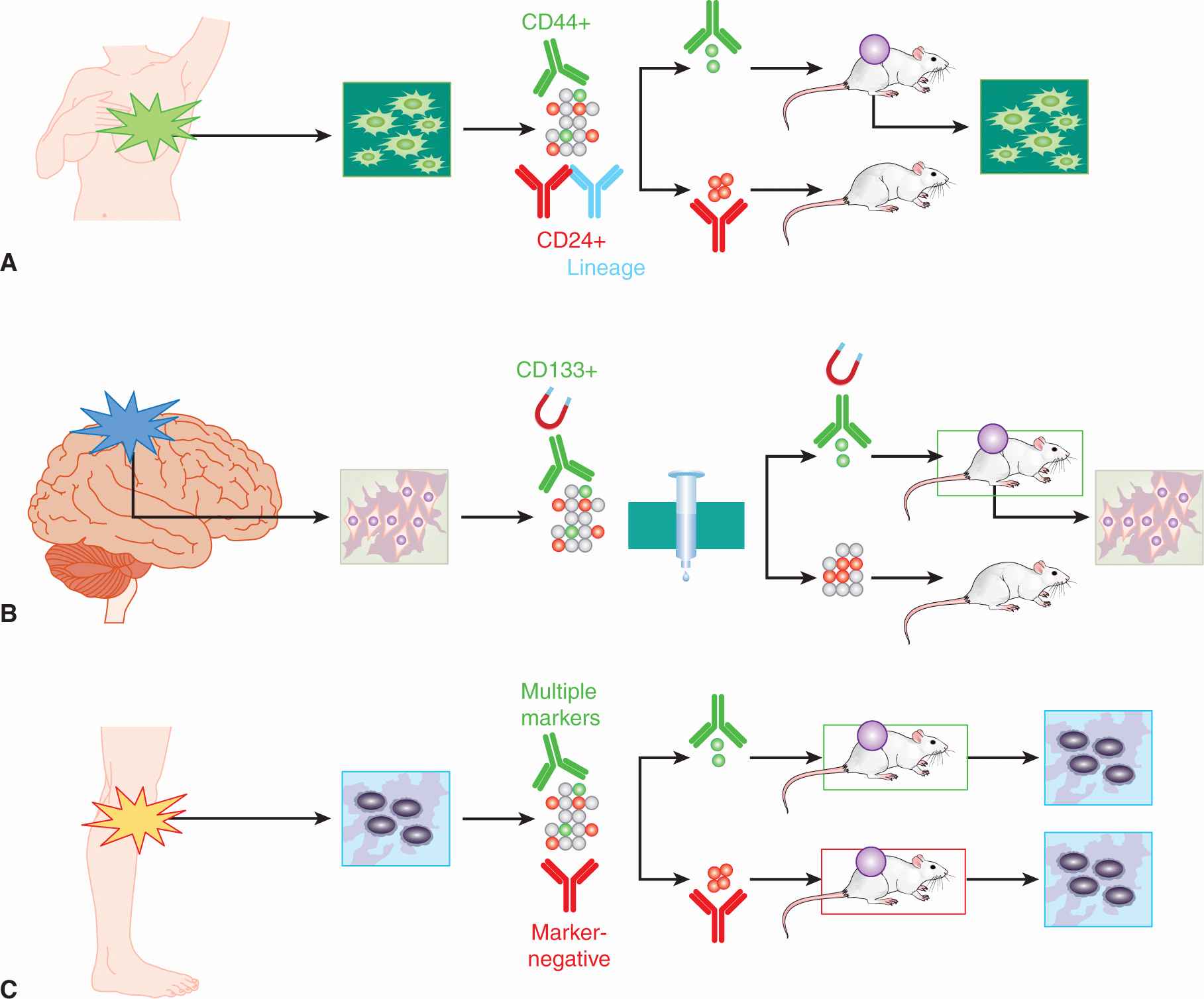
FIGURE 13–10 CSC in solid tumors. A) Lineage-negative, epithelial marker-positive, CD44+, and CD24– breast cancer stem cells can form xenografts in the mouse mammary fat pad. B) Magnetically separated CD133+ glioma stem cells orthotopically implanted into mouse brain can recapitulate human glioma xenograft complexity; CD133– cells cannot. C) Although several CSC markers had been proposed in melanoma, a systematic examination of 22 heterogenously expressed markers in melanoma failed to discover a marker that could enrich the already very frequent tumorigenic cells in melanoma; almost all malignant cells in melanoma appear to be CSCs.
13.4.5.1 Breast Cancer Breast CSCs were the first to be identified in a solid malignancy (Al-Hajj et al, 2003). Cells from 8 patients with metastatic pleural effusions and from 1 primary breast cancer were either FACS sorted or passaged through mice and sorted from xenografts. These sorted cell populations were orthotopically implanted in the mammary fat pad of female, estrogen-supplemented NOD/SCID mice that had been further immunocompromised by treatment with etoposide. The authors describe 3 series of experiments, refining the selected cell population at each step. Initially, the authors separated cells on the basis of CD44, CD24, and epithelial cell adhesion molecule (EpCAM), showing that CD44+, EpCAM+, and CD24– cell populations were all more tumorigenic than CD44–, EpCAM–, and CD24+ populations at cell doses of 2 to 8 × 105 cells. Dead cells were excluded with a fluorescent-soluble viability dye. Mouse cells in xenografted tumors were depleted by using anti-H2Kd (mouse major histocompatibility class) antibody, and the populations were enriched for tumor cells by sorting and discarding cells expressing “lineage” markers such as CD2, CD3, CD10, CD16, CD18, CD31, CD64, and CD140b, which are expressed on lymphocytes, NK cells, monocytes, B cells, endothelial cells, and fibroblasts. Within the remaining “lineage-negative” cells a CD44+/CD24– population was found in all tumors, ranging from 11% to 35% of cells (median: 15%) that was highly enriched (10- to 50-fold) for tumor-forming ability versus CD44+/CD24+/– cells (Fig. 13–10A). In a final round of purification in 3 tumors, the EpCAM+/CD44+/CD24–/Lineage– cell population (0.5% to 2.5% of all cancer cells) was found to be enriched more than 50-fold for tumor-forming cells, and as few as 200 cells could form a tumor when injected into the immunocompromised mice.
The authors demonstrated that tumor-forming potential was not related to cell cycle (ruling out brisker proliferation), and showed that tumors derived from this highly selected subpopulation gave rise to a heterogeneous xenograft with similar characteristics and cellular markers to the primary human tumor. Finally, the authors demonstrated in vivo self-renewal of CD44+/CD24–/Lineage– tumor-forming cells from 3 tumors that were able to be serially transplanted through multiple mice, yielding similar heterogeneous xenografts at each passage with no evidence of decreased ability to generate tumors.
13.4.5.2 Brain Tumors CSCs were next characterized in the brain tumors, glioblastoma and medulloblastoma (Singh et al, 2003; Singh et al, 2004). Freshly excised tumors from adults and children were cultured in chemically defined serumfree neural stem cell (NSC) media (Reynolds and Weiss, 1992); all tumors grew nonadherently as neurosphere-like clusters, and the rate of sphere formation and subsequent proliferation was proportional to the grade of the tumor. In vitro self-renewal was measured by assessing the rate of secondary sphere formation in a limiting dilution assay; clonogenicity, the ability to form a new colony, again matched tumor grade. Karyotyping revealed that sphere-forming cells had an abnormal phenotype, thus excluding contaminating NSCs. CD133 was selected as a putative marker for brain CSCs as it had been used as a marker to select normal brain stem cells (Uchida et al, 2000). CD133+ cells were isolated from glioblastoma serum-free nonadherent cell cultures using magnetic bead separation (Fig. 13–10B). CD133+ cells were highly clonogenic compared to CD133– cells, and showed higher proliferation and sphere formation at limiting dilutions of cells. In primary tumor specimens, CD133+ cells accounted for between 6% and 45% of all cells. Sphere-forming cells were exposed briefly to fetal calf serum to induce differentiation, and the phenotype of each cell line roughly matched the differentiation profile of the primary tumor, suggesting that the CD133+ stem-like cells within these glioblastoma and medulloblastoma serum-free cell lines could differentiate to recapitulate the phenotype of the original patient’s tumor.
In subsequent work CD133+ (5.7% to 28.8%) and CD133– cells isolated directly from freshly excised human medulloblastomas and glioblastomas were injected orthotopically into the brains of NOD/SCID mice at decreasing doses of cells (ie, at limiting dilution). Tumors developed in 16 of 19 mice injected with CD133+ cells, some from injection of as few as 100 CD133+ cells. Injections of up to 100,000 CD133– cells did not form tumors. Viable CD133– cells could be detected at the injection sites by species-specific pan-centromeric probes, showing that CD133– cancer cells could survive but not form tumors. The tumors arising from the CD133+ cell injections were excised and examined by immunohistochemistry, showing close resemblance to the primary patient’s tumor on the basis of immunophenotype and percentage of CD133 expression. These resected xenografts also showed reproducible karyotypes that were similar to the primary tumors. Taken together these data indicate that these xenografts were derived from the injected human CD133+ cells and had undergone differentiation in vivo to recapitulate the original tumor phenotype. Finally, self-renewal of CD133+ cells was demonstrated in vivo by excision of the tumors derived from CD133+ cells, sorting CD133+ and CD133– cells and injecting these into secondary NOD/SCID-recipient mice. As few as 100 CD133+ cells could regenerate a tumor similar to the primary tumor, whereas CD133– cells could not.
Other studies have indicated that CD133 may not mark a CSC population within every glioblastoma. Two groups reported on glioma cell lines established in serum-free stem cell media showing that some glioma lines contained CD133+ cells, while other lines were completely negative for CD133 expression (Beier et al, 2007; Gunther et al, 2008). CD133– glioma lines tended to grow more slowly in vitro and in vivo and had a different gene expression profile compared to CD133+ glioma lines. Thus the absence of a putative CSC marker in some cancers does not immediately invalidate the CSC hypothesis, but rather suggests that just as different patients’ tumors show different genotypes, they can also demonstrate different patterns of epigenetic heterogeneity. In mouse models of lung cancer, it appears that genetic and epigenetic heterogeneity are intimately linked, where the mouse stem cell marker Sca-1 marked tumor-propagating cells in K-ras/p53 mutant mouse tumors, but not in K-ras mutant tumors, while in EGFR mutant tumors, only the Sca-1-negative cells could form new tumors (Curtis et al, 2010). Finally, there is recent evidence that reexpression of “stemness” programs by reexpression of genes promoting self-renewal such as MYC, OCT4, and SOX2, may induce a degree of reprogramming stress that can lead to increased mutation rates in vitro (Ji et al, 2012). Mathematical modeling also suggests that the presence of a “CSC” population within human tumors allows them to evolve more rapidly, lending them a competitive advantage (Naugler, 2010). These observations hint at the interrelatedness of genetic and epi-genetic heterogeneities in human cancers.
13.4.5.3 Melanoma The first isolation of CSCs from patient samples of melanoma was performed using the CD133 and ABCB5 cell-surface markers (Schatton et al, 2008). Melanomas were found to express more ABCB5 than benign melanocytic nevi, thick primary melanomas expressed more than thin primary melanomas, and melanomas metastatic to lymph nodes expressed more ABCB5 than primary lesions, thus suggesting that ABCB5 is a molecular marker of melanoma progression (Kim et al, 2006). ABCB5 staining was found in all 7 fresh melanoma biopsies that were transplanted to form xenografts, and ABCB5 tended to stain areas of xenografts with less melanin. In a limited number of melanoma samples, malignant melanoma-initiating cells (MMICs) marked by ABCB5 expression were demonstrated to be a rare subpopulation in NOD/SCID mice (CSC frequency of ~1/111,000, compared to a tumor-forming frequency of 1 in ~1,000,000 unsorted melanoma cells).
In contrast to the above work, studies by Morrison and colleagues have challenged the existence of melanoma CSCs and the overall relevance of the CSC hypothesis. This group performed similar xenograft experiments in melanoma with 2 additional refinements: they injected melanoma cells mixed with high-concentration Matrigel into more severely immunocompromised mice, the NSG strain that entirely lacks NK cell activity (compared to the NOD/SCID strain in which low NK cell activity is present) (Quintana et al, 2008). The rationale was that these conditions would be most permissive for melanoma xenograft formation. This group found that tumor-forming cells in melanoma were common, such that around 1 in 4 unselected melanoma cells implanted subcutaneously could form a xenograft, and some xeno-grafts were generated after injecting a single melanoma cell per mouse. In further experiments, they demonstrated that melanoma cells sorted for a large panel of markers, such as ABCB5 and CD133, showed equivalent ability to grow into heterogeneous xenografts, expressing both marker-positive and marker-negative cells (Fig. 3–10C; Quintana et al, 2010). Thus no selection by a particular marker, no hierarchy, and thus no “CSC” could be demonstrated in melanoma. Initial experiments used cells from multiply passaged xenografts or from advanced stages III and IV melanoma patients, but this pattern of high tumor-forming ability (the frequency of TICs was ~30%) was maintained even when using single unselected cells isolated from patients with stage II melanomas (Quintana et al, 2010). Thus with sufficient optimization of the xenotransplant model, tumor-forming cells in melanoma were found to be common.
13.4.6 Criticisms of the Cancer Stem Cell Hypothesis
The observation that a xenograft could grow from almost any single unselected melanoma cell led to controversy regarding the validity of the CSC hypothesis, but, in turn, this has focused experimental design and helped drive the field forward. Criticisms of the CSC hypothesis have been discussed in detail elsewhere (Gedye et al, 2011), and can be divided into conceptual, technical, and analytical arguments. First, the concept of a normal stem cell hierarchy, and by extension a CSC hierarchy, has been challenged by Lander (2009), who pointed out that complex, seemingly hierarchical phenomena can arise from simple rules. The flocking of birds is an excellent example, where only 3 rules specifying separation, direction, and speed are needed to model this complex behavior. Observers also suffer from the cognitive bias of tending to see ordered patterns in random data. Second, there are technical criticisms of the experiments that support the CSC hypothesis, although addressing them is helping to advance the field (Fig. 13–11). For example, after the use of Matrigel and NSG mice in melanoma, many groups reevaluated their models by transplanting tumor cells into NSG mice and found that TICs remain rare in epithelial cancers (Ishizawa et al, 2010) and leukemia (Vargaftig et al, 2012). Other technical objections include the loss of CSC marker expression during tumor dissociation, failure to account for stromal cells such as fibroblasts, and the inter-species difference in tumor microenvironment between mice and humans. Finally, the analysis of CSC experiments has been criticized because mathematical analysis shows that the absolute number of CSCs appears to vary depending upon the context; for example, in some datasets, the absolute number of CSCs in the “negative” cell population outweighs the number in the selected “CSC marker-positive” fraction (Hill 2006; Stewart et al, 2011).
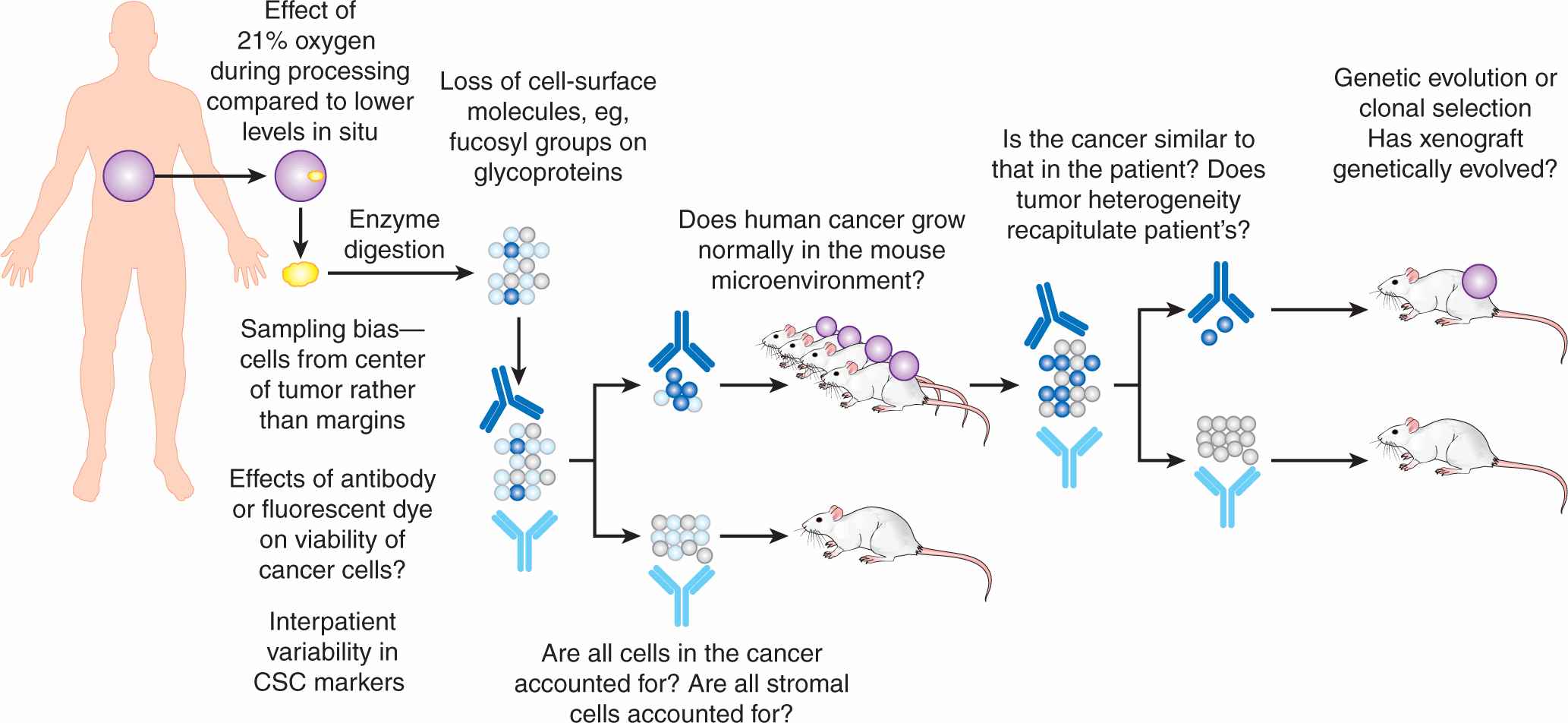
FIGURE 13–11 Technical criticisms of the CSC hypothesis. The “gold-standard” CSC assay of serial xenotransplantation has a number of technical challenges that may introduce bias into observations. Some of these challenges are unavoidable (eg, sampling at the tumor margin may not be clinically appropriate), but many are testable (eg, identifying novel markers for stromal cells, humanizing the murine microenvironment). Addressing these challenges will advance our understanding of tumor heterogeneity, and may or may not support the CSC hypothesis.
An alternative model for epigenetic heterogeneity is the concept of phenotypic plasticity (Scheel and Weinberg, 2011), where cancer cell populations are in a dynamic equilibrium controlled by epigenetic mechanisms that allow the cells to switch between different cell phenotypes in response to microenvironmental stimuli. For example, in 2 recent publications studying melanoma and non–small cell lung cancer cell lines, the histone demethylases JARID1A and JARID1B were found to be expressed in a small proportion of cells at any one time (Roesch et al, 2010; Sharma et al, 2010). In melanoma cell lines, KDM5B (also known as JARID1B) was expressed in slow cycling cells lacking expression of the proliferation marker Ki-67, but that showed evidence of stem cell-like properties, such as dye exclusion, label retention, and self-renewal. Knockdown of JARID1B was initially associated with an increase in proliferation, followed by eventual exhaustion of cell growth, suggesting that JARID1B+ cells were necessary for maintenance of the cell line. The expression of JARID1B was not hierarchal; JARID1B+ cells could be derived from JARID1B-negative cells suggesting a dynamic plasticity between cell phenotypes rather than a CSC structure.
In parallel, Sharma et al (2010) found that in cancer cell lines exposed to an EGFR tyrosine kinase inhibitor, gefitinib (see Chap. 17; Sec. 17.3.1), that although most cancer cells rapidly died, a tiny (~0.3%) quiescent subpopulation of “drug-tolerant persister” (DTP) cells survived and gradually began to proliferate. Importantly, cell lines could become drug-sensitive again after growth in the absence of drug, suggesting plasticity in the drug-tolerant phenotype. DTP cells expressed high levels of the histone demethylase KDM5A (also known as JARID1A) and in some cell lines also expressed the CSC marker CD133. Knockdown of KDM5A in these cell lines prevented the epigenetic mechanism of chromatin remodeling and modification, and completely abrogated the survival of the DTP cells. Inhibition of chromatin remodeling by inhibition of histone deacetylation (by the histone deacetylase inhibitor SAHA (suberoylanilide hydroxamic acid), or by upstream inhibition of insulin-like growth factor signaling) prevent DTP cells from surviving.
Finally, a recent examination of cell phenotypes in genomically normal embryonic stem cell cultures showed the presence of embryonic stem (ES) cells in a so-called super state, where a small (~0.2% to 1.5%) fraction of cells were “totipotent” rather than simply pluripotent (Macfarlan et al, 2012). By fluorescent cell tracking, it could be seen that all cells within the culture had the potential to express this phenotype. Modification of histones, for example, by methylation of histone 3 lysine 4 (H3K4) and acetylation of H3 and H4, was shown to partially control this process, again demonstrating an epigenetic basis for this phenotypic plasticity.
There are, therefore, several competing models to account for epigenetic heterogeneity in normal and malignant tissues. Most biological models have uncertainties and exceptions, and rather than one model being “right” or “wrong,” perhaps the CSC hypothesis and EMT models may be interrelated, though the evidence for each model has been approached from different experimental directions. This interrelationship can be exemplified by studies examining CD44+/CD24– cells in breast cancer. CD44+/CD24– breast cancer cells were described initially as breast CSCs (Al-Hajj et al, 2003). Conversely breast cancer cells that were found to be resistant to hormonal therapy (Creighton et al, 2009), chemotherapy (Li et al, 2008), or radiation (Lagadec et al, 2010), that were immunoevasive (Reim et al, 2009) or had undergone EMT (Mani et al, 2008) were found subsequently to express this same cell surface CD44+/CD24– phenotype. EMT and CSC may thus be 2 sides of the same coin (Fig. 13–12), and the mechanisms of histone modification identified in models of phenotypic plasticity might provide a mechanistic model underpinning epigenetic heterogeneity.
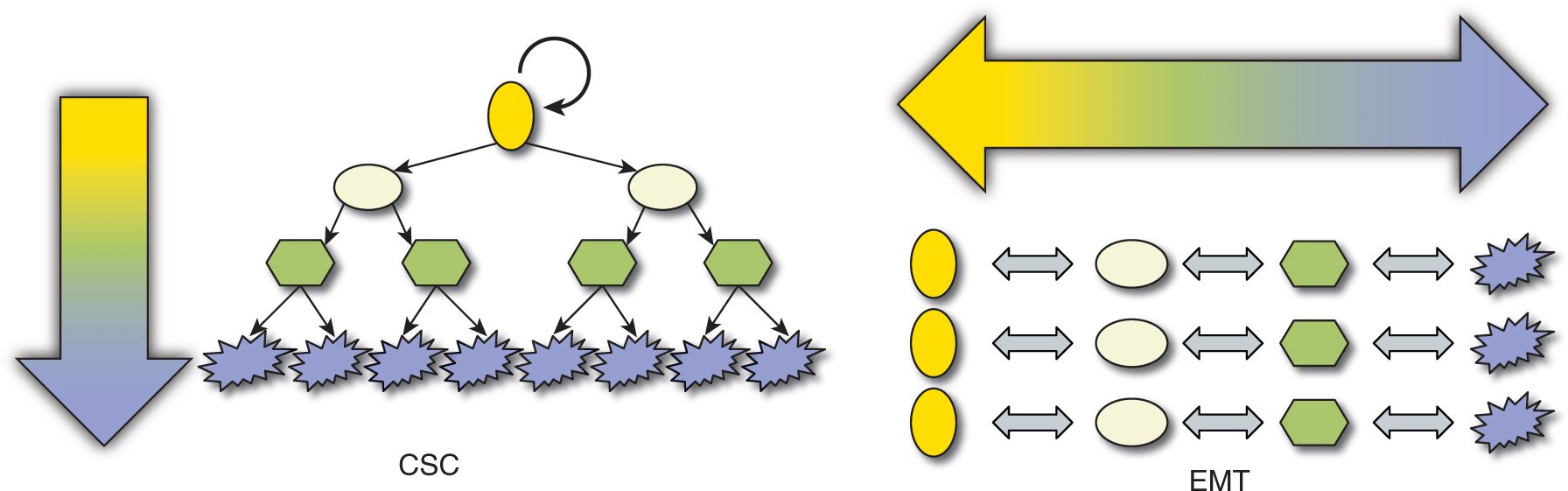
FIGURE 13–12 CSC and EMT may be 2 sides of the same coin. Increasing experimental evidence suggests considerable overlap in the cell-surface markers and biological pathways involved. The main difference remains the irreversibility of the hierarchical CSC hypothesis, where stem-like cells proliferate to terminally differentiated postmitotic progeny, whereas there is reversibility implied in the EMT model; cells with a migratory mesenchymal phenotype may take on an epithelial phenotype under the appropriate environmental stimuli.
13.4.7 Clinical Evidence Supporting the Cancer Stem Cell Hypothesis
Although both the EMT (see Sec. 13.3 and Chap. 10; Sec. 10.5.6) and CSC models can be critiqued at many levels, the true relevance of a biological model lies in its ability to improve understanding of cancer biology and clinical management. A growing body of clinical evidence supports the CSC hypothesis, including the association of CSC phenotypes with clinical outcome and with mechanisms of resistance to therapy. The novel insights afforded by a better understanding of epigenetic heterogeneity are leading to innovations in cancer therapy that are being tested in the clinic.
The CSC phenotype correlates with markers of patient outcome such as progression, survival, and distant metastasis. This has been documented from examination of the expression of single markers and from multiplex datasets.
For example, increased expression of the glioblastoma stem cell marker CD133 predicted poor patient survival (Kong et al, 2008). Multiple studies also show an association between higher CD133 expression and increased risk of progression, poorer survival and poorer response to therapy in colorectal carcinoma (Sanders and Majumdar, 2011).
Liu et al (2007) compared the gene-expression profile of CD44+/CD24– breast CSCs with that of normal breast epithelium, deriving a 186-gene “invasiveness” gene signature (IGS) that was independently and significantly associated with both overall and metastasis-free survival in women with breast cancer. Intriguingly the IGS was also associated with prognosis in medulloblastoma, lung cancer, and prostate cancer, suggesting that this signature is relevant to other cancers. Gene expression profiling of functionally validated leukemic stem cell populations yielded a signature of “stemness” associated with poor prognosis (Eppert et al, 2011). In this study, multiple patients’ samples were engrafted in mice, but showed different phenotypes of leukemic stem cell subsets (eg, CD34+, CD34+/CD38–). Despite these differences in marker expression, a core gene expression signature was present in the engrafting leukemic stem cell subset.
Pece et al (2010) isolated slow-cycling mammary gland stem cells in mammosphere culture from normal human breast biopsies, using a fluorescent dye that is retained by nondividing cells. Gene expression array analysis compared dividing (nonfluorescent) versus nondividing (fluorescent) cells, identifying a putative signature of human mammary stem cells. This gene signature of slow-cycling cells identified cell markers such as CD49f, Delta and Notch-like EGF-related receptor (DNER), and delta-like-1 (DLL1), which could be used to isolate prospectively normal mammary stem cells from mammosphere cultures. The gene signature was also able to predict tumor grade in breast carcinoma; high grade 3 tumors highly expressed the mammary stem cell signature compared with low grade 1 tumors.
Additionally cancer cells expressing markers such as CD49f, DNER and DLL1 were much more frequent in grade 3 tumors. A gene expression signature of normal intestinal stem cells in mice, defined by ephrin-B2+ cells, was shown to be predictive of poor clinical outcome in human colorectal carcinoma (Merlos-Suarez et al, 2011). Also, in a recent study using single-cell PCR (see Chap. 2, Sec. 2.2.5) from individual colonic cells, gene expression signatures could be defined for different normal tissue compartments, such as enterocytes, goblet cells, and cells from the top (cells that are proliferating and differentiating) and bottom (where cells are more quiescent and “stem-like”) of the crypt (Dalerba et al, 2011). The gene signature of these terminally differentiated top-of-crypt cells was associated with improved prognosis in patients with colorectal carcinoma. In addition, colorectal cancer xenografts were generated from decreasing numbers of injected EpCAM+/CD44+ colorectal CSC, including 1 xenograft derived from a single EpCAM+/CD44+ colorectal CSC. This clonally derived tumor recapitulated almost perfectly the heterogeneity observed in the patient’s tumor, providing further evidence in support of the CSC hierarchy within colorectal carcinoma.
Patterns of failure of cancer therapy are also consistent with the CSC hypothesis. Surgery is important in the treatment of most solid malignancies, and surgical margin status is correlated with local control of disease and long-term survival in many tumor types, including head and neck cancer. Some tumors recur at the site of resection despite apparently adequate margins; Prince et al (2007) demonstrated that the CD44+/BMI1+ CSCs detected in head and neck squamous carcinoma are most often positioned in a peristromal location at the leading edge of the tumor, abutting connective tissue, and may be the cause of recurrence. Calabrese et al (2007) showed that glioblastoma stem cells are located in a perivascular location, perhaps accounting for the propensity of glioma to extend beyond the apparent tumor margin, leading to frequent disease relapse following attempted curative surgery.
Some types of resistance to chemotherapy can be accounted for by the CSC hypothesis (see Chap. 19, Sec. 19.3.5). Liu et al (2006) showed that CD133+ CSC in de novo glioblastoma cell lines are intrinsically more chemoresistant than more differentiated cells; Li et al (2008) showed intrinsically resistant breast CSC; and Dylla et al (2008) showed that colorectal CSCs are more likely to survive treatment with oxaliplatin chemotherapy. Finally, in del(5q) myelodysplasia, a subpopulation of quiescent, CD34+, CD38–/low, and CD90+ myelodysplastic stem cells was found to persist and eventually cause treatment failure in patients who had clinical complete remissions after treatment with lenalidomide (Tehranchi et al, 2010).
Resistance to radiation treatment has also been shown to be an intrinsic property of human glioblastoma CSC (Bao, Wu, McLendon et al, 2006) and breast CSC (Diehn and Clarke, 2006). The mechanism of radioresistance of CSCs may be a result of lower levels of reactive oxygen species compared to more differentiated cancer cells, which allows CSCs to mitigate DNA damage caused by radiotherapy (Diehn et al, 2009; Brunner et al, 2012).
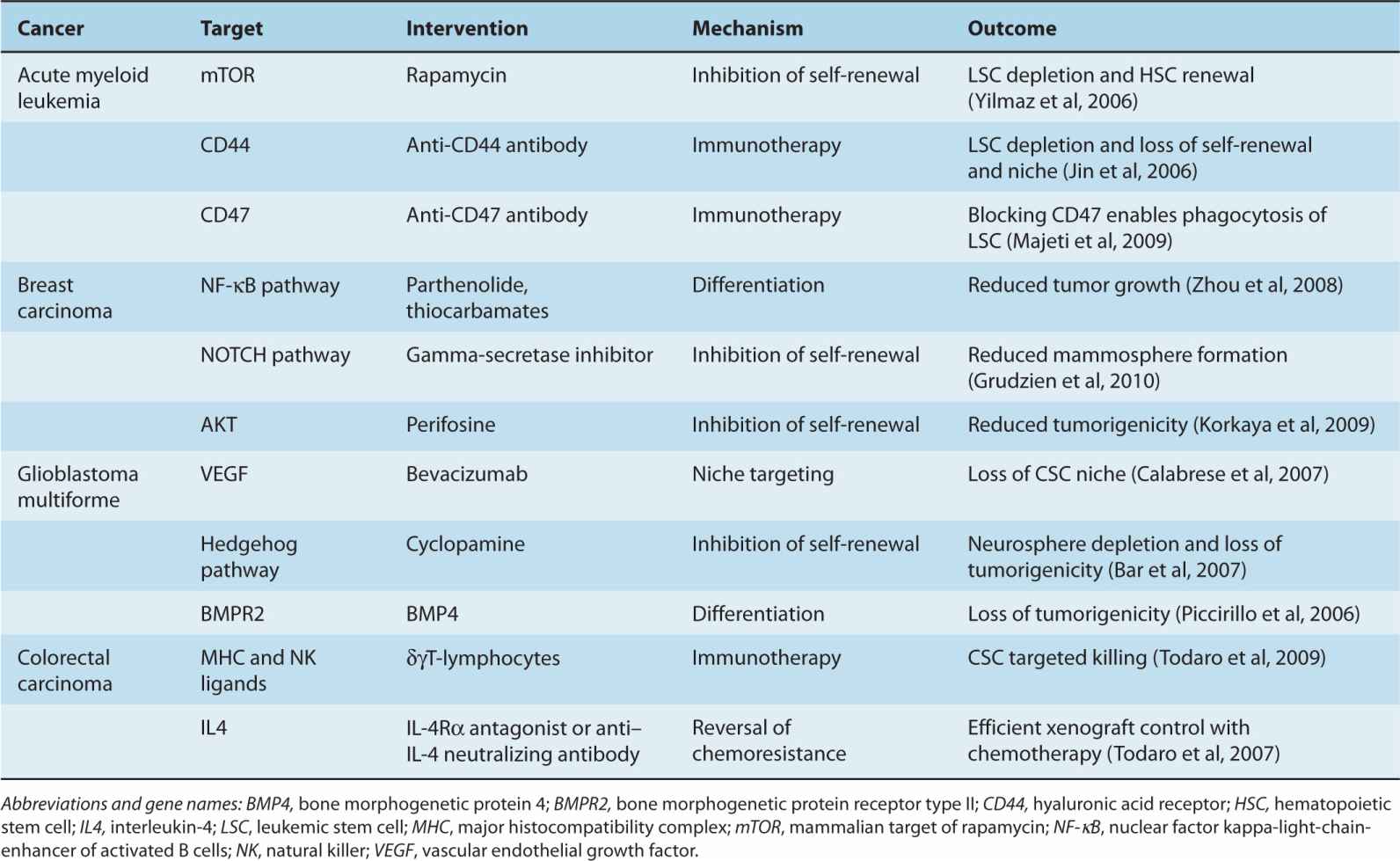
13.5 TARGETING CANCER STEM CELLS
The CSC hypothesis is informing advances in cancer therapy, although it is unlikely that cancer would be adequately treated by attempting to eliminate CSCs alone, as more differentiated cancer cells, such as “progenitor” or “transit-amplifying” cells, are likely to retain sufficient proliferative potential to cause patient death. Rather, targeting CSCs is likely to be useful as an adjunct to standard therapies that debulk the tumor, such as surgery, radiotherapy, and chemotherapy. CSC-focused therapies may be most useful in the neoadjuvant and adjuvant settings, in conjunction with attempted curative treatment of primary disease (Table 13–2).
TABLE 13–2 Targeting cancer stem cells.
13.5.1 Differentiation Therapy
One implication of the CSC hypothesis is that cotreatment with agents that cause differentiation of quiescent stem-like cancer cells to cycling progenitor/transit-amplifying cells might make them more susceptible to therapy. A clinical example of this phenomenon is the use of all-trans-retinoic acid (ATRA) and arsenic trioxide in the treatment of M3 acute promyelocytic leukemia (APML). ATRA and arsenic trioxide act by different mechanisms to destabilize the PML-RARα fusion translocation oncogene which is the driver event in most cases of APML. These agents cause differentiation and loss of self-renewal in the leukemic promyelocytes, and are so effective when combined that clinical trials have shown durable complete remissions in patients without the need for cytotoxic chemotherapy (Park and Tallman, 2011).
Efforts to overcome resistance to chemotherapy associated with CSCs in other cancers are being tested in preclinical models and entering early phase clinical trials (see Chap. 19, Sec. 19.3.5). For example, parthenolide, a natural product derived from the traditional medicinal herb feverfew that inhibits the nuclear factor kappa B (NF-κB) pathway, has been shown to induce apoptosis in LSCs while sparing normal hematopoiesis in xenograft models of AML (Guzman et al, 2007). NOTCH4 has been shown to regulate breast CSCs and γ-secretase inhibitors of the NOTCH pathway (see Chap. 8, Sec. 8.4.2), are in early phase clinical trials in breast cancer (McDermott and Wicha, 2010). In another preclinical example, Gupta et al (2009) used high-throughput drug screening to identify salinomycin as a selective inhibitor of CD44+/CD24– breast cancer stem-like cells in breast cancer cell lines.
The combination of cyclopamine, rapamycin, and gemcitabine was shown to be effective against primary human pancreatic carcinoma xenografts, whereas any combination of 2 agents was no more effective than gemcitabine alone (Mueller et al, 2009). Cyclopamine is a natural product derived from the corn lily that inhibits the Hedgehog pathway (see Chap. 8, Sec. 8.4.3), and in pancreatic carcinoma, this signaling pathway was shown to be critical to CSC self-renewal, migration, and proliferation. Cyclopamine has proven toxic in humans, but a number of synthetic inhibitors of the Hedgehog pathway are in clinical trials in basal cell carcinoma, pancreatic carcinoma, medulloblastoma, head and neck squamous carcinoma, and non–small cell lung cancer (Clayton and Mousa, 2011).
13.5.2 Immunological Targeting of Cancer Stem Cells
Attempts to improve or induce immunological surveillance, recognition, and destruction of residual CSC after debulking with chemotherapy, radiation, or surgery provide a rational approach to try to improve outcome for patients (see Chap. 21, Sec. 21.5). For example, Cioffi et al (2012) demonstrated the potential of a bispecific antibody in targeting pancreatic CSCs, showing that cytotoxic CD8+ T lymphocytes could be “redirected” against primary human pancreatic xenografts. The bispecific antibody, MT110, is synthesized to recognize 2 different antigens; one F(ab) region recognizes CD3, a pan-lymphocyte marker, and the other recognizes EpCAM, a molecule highly expressed on CSCs in pancreatic cancer. The CSC subpopulation within these tumors was not resistant to this treatment, and an improvement in survival was seen in a mouse xenograft model. Phase I clinical trials suggest evidence of activity, but also led to dose-limiting diarrhea and raised liver enzymes in humans. Cellular immune responses to CSCs have also been investigated, for example, through a γδT-cell response to colorectal CSCs (Todaro et al, 2009). Recognition and killing of colorectal CSCs was augmented by pretreatment with zoledronate to upregulate isoprenoids on the surface of CSCs. Finally, vaccination against antigens expressed on stem cells or CSCs shows some pre-clinical evidence of activity in targeting CSCs. Vaccination of immunocompetent mice with murine ES cells generated a robust immune response that rendered these mice strongly resistant to subsequent engraftment with a murine lung cancer cell line. This was not a result of an allogeneic immune response, as vaccination with syngeneic embryonic fibroblasts failed to generate similar protection (Yaddanapudi et al, 2012). Ning et al (2012) employed an analogous experimental plan, but vaccinated immunocompetent mice with stem-like cancer cells from murine cancer cell lines. Mice were immunized with ALDH+ stem-like cells isolated from cell lines prior to inoculation with the cancer cell line itself (Ma and Allan, 2011). Immunized mice showed in vivo and in vitro immune responses that also slowed tumor progression. There are many complexities and challenges in active cancer immunotherapy (Cebon et al, 2007) but human studies of active vaccination against CSC have commenced.
13.5.3 Targeting the Cancer Stem Cell Niche
Somatic and ES cells can only exist in a tightly regulated stem cell niche, with bidirectional signaling to maintain their phenotype, which, in turn, maintains the cells required for their niche support (Moore and Lemischka, 2006). Evolving evidence suggests that CSCs may also exist within, and signal to maintain, a functional niche, providing another potential target for cancer treatment. Bao, Wu, Sathornsumetee et al (2006) found that vascular endothelial growth factor (VEGF) was highly expressed by CD133+-derived glioma stem cell cultures and xenografts compared with matched CD133– cells. This secretion was further enhanced under hypoxic culture conditions. When CD133+ glioma stem cell-conditioned media was applied to endothelial cell (EC) cultures, it significantly enhanced EC migration and tube formation. The anti-VEGF antibody bevacizumab abrogated these proangiogenic effects and also suppressed tumor xeno-graft formation.
Calabrese et al (2007) demonstrated that primary brain cancers exist within and exert control over vascular niches that allow them to maintain the CSC subpopulation. Using multiphoton laser-scanning immunofluorescence microscopy to digitally reconstruct tumor sections, nestin-positive cells were found to colocate with capillaries in 4 glioma biopsies. In vitro experiments showed that CD133+ brain CSC interact with ECs in coculture, and, in turn, these ECs were able to maintain and propagate the CD133+ fraction of tumors. Finally, the authors demonstrated that ECs promoted tumor formation of CD133+ brain CSCs in orthotopic xeno-grafts, and that disruption of the vascular niche by erlotinib and the antiangiogenic agent bevacizumab directly ablated the self-renewing CSC population and inhibited xenograft growth (Calabrese et al, 2007).
Several recent publications suggest that glioblastoma stem cells can contribute directly to the tumor microenvironment by differentiating into tumor ECs. In human tumors (Ricci-Vitiani et al, 2010; Wang, Chadalavada et al, 2010) and in lentiviral lineage-restricted Cre-Lox mouse models (Soda et al, 2011), which permit the introduction of oncogenes in limited numbers of cells in vivo, glioblastoma cells differentiated into CD31+CD144+ ECs, which contribute to functional blood vessels. Some of these vessels lacked VEGF receptors and were insensitive to VEGF-targeted therapies. A similar process was also described in ABCB5+ VEGFR2+ melanoma cells. This finding illustrates an additional level of complexity in the biology of CSCs in the context of their microenvironment and in the challenge faced in advancing cancer therapy.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


