Joshua T. Schiffer, Lawrence Corey
Herpes Simplex Virus
Herpes simplex viruses (HSV-1 and HSV-2) produce a wide variety of illnesses, including mucocutaneous infections, infections of the central nervous system (CNS), and an occasional infection of visceral organs; some of these conditions may be life threatening. HSV-2 is linked, both at a biologic and epidemiologic level, to increased rates of both acquisition and transmission of human immunodeficiency virus type 1 (HIV-1) infection. The availability of effective chemotherapy for HSV infection has made prompt recognition of infection important. Unfortunately, anti-HSV therapy does appear to alter the effects of recent or prior HSV-2 infection on HIV acquisition or transmission.
The word “herpes” (from the Greek, “to creep”) has been used in medicine since antiquity. Cold sores (herpes febrilis) were described by the Roman physician Herodotus in 100 ad.1 Genital herpes was first described by John Astruc, physician to the king of France in 1736: the first English translation appeared in his treatise on venereal disease in 1754.2,3 Infection in orolabial lesions was transmitted to other humans in the late 19th century. The disease was successfully transferred to rabbits in the early 20th century, and HSV was grown in vitro in 1925.4,5 In the early 1960s, Schneeweiss and Dowdle and Nahmias reported two antigenic types of HSV with different sites of viral recovery.6
Description of the Virus
Structure
The eight known human herpesviruses (HHVs) are divided by genomic and biologic behavior into three groups: the alphaherpesviruses (HSV-1, HSV-2, and varicella-zoster [VZV]), the betaherpesviruses (cytomegalovirus, HHV-6, HHV-7), and the gammaherpesviruses (Epstein-Barr virus, Kaposi sarcoma–associated herpesvirus [KSHV], or HHV-8). Herpesviruses are morphologically similar, possessing an internal core containing double-stranded DNA, an icosahedral capsid with 162 capsomers, an amorphous material surrounding the capsid called a tegument, and a lipid envelope containing viral glycoproteins on its surface. Their overall diameter is about 160 nm.7 Despite common morphologic features, the biologic and epidemiologic features of each of the herpesviruses are different. Although HSV-1 and HSV-2 are the two most closely related herpesviruses, the two agents are serologically and genetically distinct.6
The genome of HSV is a linear, double-stranded DNA molecule (molecular weight about 1 × 108) that encodes about 90 transcriptional units, 84 of which appear to encode proteins. The genetic organization has sequences from both terminal ends of the genome repeated in an inverted fashion. This divides the genome into two unique components.8 The overall sequence homology between HSV-1 and HSV-2 is about 50%.9 The homologous sequences are distributed over the entire genome map, and most of the polypeptides specified by one viral type are antigenically related to polypeptides of the other viral type. Many type-specific regions unique to HSV-1 and HSV-2 proteins do exist, however, and many of these regions appear to be important in evading host immunity. Intertypic recombinants have also been identified.
HSV is genomically stable, and restriction endonuclease or sequence analysis of viral DNA can be used to distinguish between the two subtypes and among strains of each subtype.10,11 The variability of nucleotide sequences from clinical strains of HSV-1 and HSV-2 is such that HSV isolates obtained from two individuals can be differentiated.12 Isolates from epidemiologically related sources, such as sexual partners, mother-infant pairs, or victims of a common-source outbreak, are identical.13–15 Whole-genome sequencing has only recently been extended to a variety of clinical isolates. The degree of sequence diversity across the globe is only partially characterized.16 Although hot spots of sequence variability are increasingly being identified in specific genes, the role of this variability in HSV pathogenesis is only beginning to be evaluated.
Replication
Viral replication has nuclear and cytoplasmic phases. The initial steps of replication include attachment and fusion between the viral envelope and cell membrane to liberate the nucleocapsid into the cytoplasm of the cell. Several cellular receptors and viral envelope glycoproteins are required for viral attachment. The initial attachment to the cell membrane involves the interactions of viral glycoproteins C and B with cellular heparin sulfate.17 Subsequently, viral glycoprotein D binds to cellular co-receptors that belong to the tumor necrosis factor receptor family of proteins, the immunoglobulin superfamily (nectin family), or both.18,19 The ubiquity of these receptors underscores the wide host range of herpesviruses, and their presence on sensory neurons implicates their role in the development of neuronal infection, and therefore latency.20–22
After attachment, the de-enveloped tegument capsid structure is transported to nuclear pores where viral DNA is released into the nucleus. After fusion of the virion envelope with the host cell membrane, the virions release several functional proteins. The virion host shutoff protein shuts off synthesis (by increasing cellular RNA degradation), whereas VP16 turns on transcription of immediate early genes of HSV replication.23 Some of these immediate early gene products (designated α-genes), are important determinants of neurovirulence in animal models, whereas others are required for synthesis of a subsequent polypeptide group, the β or early polypeptides. Many β-proteins are regulatory proteins and enzymes required for DNA replication. Most antiviral drugs in current use or development interfere with β-proteins, such as the viral DNA polymerase enzyme or DNA helicase.24,25 Transcription of the viral genome, replication of viral DNA, and assembly of capsids take place in the nucleus.26 Moreover, the late (γ) class of HSV genes requires viral DNA replication for expression. These late proteins are structural and assist with viral egress. DNA replication takes place in a “rolling circle” pattern similar to a roll of toilet paper. Specific viral genes “clip” the end of the viral DNA into the procapsid.
After nucleocapsids are assembled in the nucleus, envelopment occurs as the nucleocapsids bud through the inner nuclear membrane into the perinuclear space. In some cells, viral replication in the nucleus forms two types of inclusion bodies: (1) type A basophilic Feulgen-positive bodies that contain viral DNA and (2) eosinophilic inclusion bodies that are devoid of viral nucleic acid or protein and represent a “scar” of infection. Virions are then transported through the endoplasmic reticulum and Golgi apparatus to the cell surface. The entire viral production cycle takes 4 to 6 hours, and an infected cell survives for 16 to 20 hours. HSV is cytopathic to cells that harbor the full cycle of HSV replication.27
Molecular Features of Latency
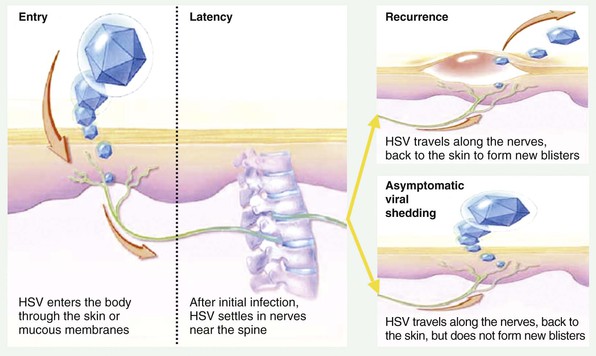
HSV infection of some neuronal cells does not result in cell death. Instead, viral genomes are maintained by the cell in a repressed state compatible with survival and normal activities of the cell, called latency.28,29 Latency is associated with transcription of only a limited number of virus-encoded RNAs.30–32 Subsequent activation of the viral genome may occur, resulting in the normal pattern of regulated viral gene expression, replication, and release of HSV, although without apparent damage to the infected neuron. The release of virions from the neuron follows a complex process of anterograde transport down the length of neuronal axons.33,34 Subsequent viral entry into epithelial cells can result in viral replication; this process is termed reactivation.35,36
Although infectious virus typically cannot be cultured from sensory or autonomic nervous system ganglia dissected from cadavers, maintenance and growth of the neural cells in tissue culture results in production of infectious virions (explantation) and in subsequent permissive infection of susceptible cells (cocultivation).37 The fact that HSV replication was first detected in neurons during reactivation in vitro suggested that the neuron harbors latent virus in vivo.30 Viral DNA and RNA have since been found in neural tissue at times when infectious virus cannot be isolated.32,38 Documentation of individual neurons infected with multiple strains of drug susceptible and resistant virus has been shown in severely immunosuppressed persons.39 More recently, this phenomenon has been reported in immunocompetent subjects,40 suggesting that the ganglia is probably continually reseeded with HSV throughout the course of chronic infection, although drug resistance is typically not of clinical importance in immunocompetent patients.
Three noncoding RNA “latency-associated” transcripts (LATs) are the only transcripts in abundance in the nuclei of latently infected neurons.30,41,42 Deletion mutants of the genomic region that can become latent have been made, and the efficiency of their later reactivation is reduced.41,43 In addition, substitution of HSV-1 LAT for HSV-2 LAT induces an HSV-1 reactivation pattern. In both trigeminal and dorsal root ganglia, LATs appear to maintain, rather than establish, latency.44 HSV-1 LATs promote the survival of acutely infected neurons perhaps by inhibiting apoptotic pathways.45,46 LAT-derived microRNA is highly expressed during latency and appears to silence expression of the key neurovirulence factor ICP34.547 and prevent expression of ICP0, an immediate early protein that is vital to HSV reactivation, through antisense binding of mRNA. However, exit from latency appears to correlate with complete derepression of HSV replication genes rather than just a small cluster of viral genes.48
Although latency is the predominant state of virus on a per neuron basis, the high frequency of oral and genital tract reactivation for HSV-1 and HSV-2 suggests that the virus is rarely quiescent within the entire biomass of ganglionic tissue.49,50 Recent experimental data suggest that HSV-2 antigen is frequently and nearly continually shed into genital mucosa. Mathematical models of HSV-2 reactivation predict that viral release from the ganglia occurs in a nearly continuous drip,50 whereas only a single HSV particle from neurons is necessary to spark viral replication in genital epithelial cells.50 Recent work employing microdissection plus real-time polymerase chain reaction (PCR) assay of individual neurons from cadaveric trigeminal ganglia explants revealed that many more neurons (2% to 10%) harbor HSV than would be predicted by in situ hybridization studies for LAT.51,52 Moreover, viral copy number is highly variable between neurons with extremely high levels in certain neurons.51,52 HSV DNA copy numbers are similar in LAT-positive and LAT-negative neurons, adding uncertainty to the role that LATs play in preventing reactivation.
LAT transcript abundance and low genome copy number correlate with subnuclear positioning of HSV genomes around the centromere.53 Indeed, chromatization of HSV DNA appears to play a vital role in silencing expression of lytic replication genes.54,55 Although certain viral transcripts are known to be necessary for reactivation from latency, the molecular mechanisms of HSV latency are not fully understood, and strategies to interrupt or maintain latency in neurons are in developmental stages.56
Epidemiology
Herpes simplex viruses have a worldwide distribution and are found in the most remote human populations. There are no known animal vectors for HSV, and although experimental animals are easily infected, humans appear to be the only natural reservoir. Herpes infection is the predominant cause of genital ulcers throughout the world. This is due to an overall decrease in Treponema pallidum and chancroid infections in most populations,57 increase in PCR usage for HSV detection, and increase in HSV-2 reactivation frequency among HSV/HIV-coinfected persons.
HSV-1
Infection with HSV-1 is acquired more frequently and earlier than infection with HSV-2. More than 90% of adults have antibodies to HSV-1 by the fifth decade of life. Prevalence of antibody to HSV-1 increases with age and demonstrates an inverse correlation with socioeconomic status. In much of Asia and Africa, HSV-1 infection is nearly universal and is acquired early in childhood. However, in post–World War II era Western populations, 80% to 100% of middle-aged adults of lower socioeconomic status had antibodies to HSV-1, compared with only 30% to 50% of adults in higher socioeconomic groups.58,59 Serosurveys continue to show a decline in the age-specific prevalence rates for HSV-1 infection in both the United States and most of Europe, although socioeconomic class distinctions remain.60 In developed countries, a decrease in HSV-1 acquisition in childhood accounts for the increased frequency of sexually acquired HSV-1 infections in adolescents, as well as the increased proportion of neonatal HSV cases that are due to HSV-1.59,61–63 Although primary HSV-1 and HSV-2 are clinically indistinguishable, in the United States, genital HSV-1 is now more common in white patients whereas primary genital HSV-2 remains more common in black patients.64
HSV-2
Antibodies to HSV-2 start to appear during puberty and correlate with initiation of sexual activity.60,65 Widespread use of serologic testing has provided a detailed characterization of a worldwide HSV-2 pandemic over the past 2 decades (Fig. 138-1).60,66,67,68 Most African surveys indicate very high levels of infection, although risk varies within each country according to gender and region.69 Seroprevalence is lower in Europe, Australia, Latin America, and Asia, although it remains highly dependent on the risk of the group being evaluated. In the United States, nationwide surveys showed an increase in HSV-2 seroprevalence from 16.4% to 21.7% of adults between 1979 and 1991, with a decrease to 17% from 1999 to 2004.60,66,70 The cumulative lifetime incidence of HSV-2 reaches 25% in white women, 20% in white men, 80% in black women, and 60% in black men. The higher rates of HSV-2 among African Americans may reflect patterns of sexual networking rather than high-risk individual behavior.71
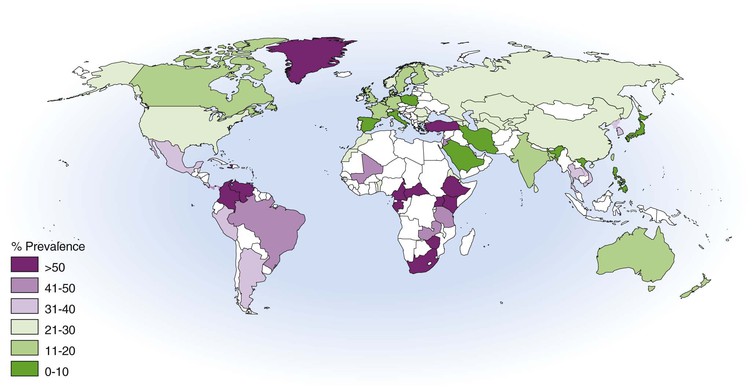
HSV-2 prevalence in a population is defined by geographic region, gender, sexual habits, and study population. There is a consistently higher prevalence of HSV-2 in women than in men,58,59,63,67 and HSV-2 infection is also prevalent among HIV-infected persons, persons recruited from sexually transmitted disease clinics, and men who have sex with men.67,72 HSV-2 antibody levels are closely related to the lifetime number of sexual partners, age of sexual debut, and a history of other sexually transmitted diseases.73,74
The global incidence of HSV-2 infection has been estimated at 23 million new cases per year.75 Local incidence rates of HSV-2 infection are often difficult to estimate owing to the common nature of asymptomatic seroconversion, attenuation of symptoms due to prior HSV-1 infection, location of lesions in nonvisible locations (e.g., perianal), and differential access to health care and diagnostics. For instance, among HSV-seronegative women in the control arm of an HSV-2 vaccine trial, only half of seroconversions were clinically symptomatic.76 Nevertheless, data are accumulating and vary between populations based on risk characteristics and geography. Vaccine and condom prevention studies conducted in serodiscordant American couples document HSV-2 seroincidence levels between 6.7 and 8.6 infections per 100 person-years for women and 1.5 to 3.7 per 100 person-years for men.77–79 Seroincidence in several high-risk urban youth cohorts was 11.7 cases per 100 person-years.80 In African populations characterized by high preexisting HSV-2 seroprevalence, seroincidence was 1.8 to 12.9 per 100 person-years, with higher acquisition rates among persons infected with HIV-1 and among seronegative women in a monogamous relationship with a seropositive man.81–83 Among adolescent cohorts in sub-Saharan Africa, incidence exceeding 20 per 100 person-years has been reported.84,85
HSV-2 Risk Factors
Cofactors for risk for genital HSV-2 acquisition in an individual are well defined from prospective trials. Despite equivalent shedding rates according to gender,86 women are consistently at higher risk for HSV-2 acquisition than men. Possible explanations include greater mucosal surface area, higher likelihood of asymptomatic ulcers in men that may facilitate transmission, or positioning within social networks. It is uncertain whether past HSV-1 infection reduces the risk for infection with HSV-2. However, persons with prior HSV-1 infection are three times as likely to acquire HSV-2 subclinically.76 In contrast to bacterial sexually transmitted diseases, HSV-2 is commonly transmitted within long-term couples rather than casual sexual relationships. Longitudinal studies of such couples showed transmission rates varying from 3% to 12% per year.79,87 The median time to transmission within discordant couples is 3 months, with a median number of only 40 sex acts prior to transmission, or an approximate 3.5% per-coital risk.79 Moreover, one third of source partners within serodiscordant couples deny a history of genital lesions.88
Knowledge of a long-term partner’s HSV-2–positive status decreases transmission incidence by 50%, highlighting the importance of formal diagnosis and disclosure of infection.79,89 Consistent condom use decreases HSV acquisition among women, and chemoprophylaxis of the source partner also decreases transmission.79,87 Circumcision decreases both HIV-1 and HSV-2 acquisition risk in men.90–92 However, male circumcision does not impact transmission rates of HSV-2 among female partners.93
Transmission
In 1921, Lipschutz inoculated material from genital herpetic lesions into the skin of humans, eliciting clinical infection within 48 to 72 hours in six persons and within 24 days in one case.5 Transmission of HSV most frequently occurs through close contact with a person who is shedding virus at a peripheral site, at a mucosal surface, or in genital or oral secretions.68,94,95 Infection occurs by inoculation of virus onto susceptible mucosal surfaces (e.g., the oropharynx, cervix, conjunctivae) or through small cracks in the skin. Because HSV is readily inactivated at room temperature and by drying, aerosol and fomitic spread are unusual. Transmission of HSV-1 from orogenital contact is increasingly recognized, perhaps because of reduction in age-specific prevalence of HSV-1 at the time of sexual debut.96 Spread of HSV-1 infection from oral secretions to other skin areas is a hazard of certain occupations (e.g., dentists, respiratory care unit personnel), and laboratory-acquired and nosocomial outbreaks in hospital or nursery personnel have been reported.97 Outbreaks among wrestlers are well recognized.98 Transmission of HSV can occur in infants born to mothers excreting HSV at delivery.99 Anal and perianal infections with HSV-1 or HSV-2 are common among sexually active populations of men who have sex with men.100 The majority of cases occur within 5 days of contact, highlighting the short incubation period of primary infection.
Precise virologic determinants of transmission likelihood are poorly understood. For HIV infection, a clear relationship between genital and plasma HIV viral load and per-coital risk for HIV transmission is established.101,102 However, because genital HSV-2 levels fluctuate extremely rapidly over hours both on and off therapy,103,104 the degree to which source partner viral load during sex impacts likelihood of transmission is unknown.105 Subclinical or asymptomatic shedding of HSV in oral and genital secretions is a hallmark of infection,68,86,106 even in immunocompromised persons,107 and transmission occurs more commonly during asymptomatic shedding.
Frequency of detectable shedding is markedly heterogeneous among those seropositive for HSV-2, likely influencing the variability in per-coital transmission rates that have been reported.106 A modeling study predicted that a core group of “super spreaders” with high reactivation rates could account for a disproportionately large percentage of new infections.108 DNA polymerase inhibitors that decrease frequency of asymptomatic shedding as well as peak HSV-2 titers during recurrence also decrease transmission within serodiscordant couples.87 Yet frequency of symptomatic recurrences poorly predicted likelihood of transmission in a prospective trial, again highlighting the high transmission risk for asymptomatic shedding.109 Prolonged asymptomatic shedding episodes lasting several days have been well described even among immunocompetent patients taking daily antiviral therapy.86,104
Pathogenesis
Primary Infection
Exposure to HSV at mucosal surfaces or abraded skin sites permits entry of the virus and initiation of its replication in cells of the epidermis and dermis.110 Initial HSV infection is often subclinical, without apparent lesions. In animal models and human subjects, both clinical acquisition and subclinical acquisition are associated with sufficient viral replication to permit infection of either sensory or autonomic nerve endings, with the subsequent potential for lifelong reactivation patterns.32,49,110,111 After traversing the neuroepithelial gap and entering into the neuronal cell, the virus or nucleocapsid is transported intra-axonally to the nerve cell bodies in ganglia.112 For HSV-1 infection, trigeminal ganglia are most commonly infected, although extension to the inferior and superior cervical ganglia also occurs.28,29 With genital infection, sacral nerve root ganglia (S2 to S5) are most commonly affected.35 Recent data suggest a possible role for autonomic tissue in maintenance of latent infection.113 In humans, the interval from inoculation of virus in peripheral tissue to spread to the ganglia is unknown.
High-level viral replication occurs in ganglia and contiguous neural tissue during primary infection, although lytic transcripts are detectable during reactivations in mouse and guinea pig models as well.110,111,114 After initial inoculation of the neural ganglion, virus spreads to other mucosal skin surfaces by centrifugal migration of infectious virions through peripheral sensory nerves. This mode of spread explains the characteristic development of new lesions distant from the initial crop of vesicles in patients with primary genital or orolabial HSV infection, the large surface area over which these vesicles may be visualized, and the recovery of virus from neural tissue distant from neurons innervating the inoculation site.115 Contiguous spread of virus is also likely to take place via autoinoculation and allow further extension of disease.103 Viremia is present during approximately 25% of primary HSV-2 infections, and its presence may affect the natural history of HSV-2 disease in terms of site, severity, and frequency of reactivation.116
Ganglionic Immunity
After resolution of primary disease, viral DNA is present in 2% to 11% of ganglion cells in the anatomic region of initial infection.51 Therefore, many neurons may contribute to reactivation. Aside from the previously described molecular features of latency, host T-cell responses at the ganglion level may influence the frequency and severity of HSV reactivation. Although it is not known whether reactivating stimuli transiently suppress these immune cells or independently upregulate transcription of lytic genes, or both, it is well documented that CD8+ T cells juxtapose to HSV-1 latently infected neurons in human trigeminal ganglia.117 These activated cells are reactive to a broad array of HSV antigens.118 In murine models, resident CD8+ lymphocytes and lymphocyte-derived cytokines appear to be important in preventing infectious virions from being transported down the length of the axon for release in the basal layer of the epidermis119–121 and can block reactivation both with interferon-γ release122 and granzyme B degradation of immediate early protein ICP4.123 In addition, there appears to be a latent viral load in the entire biomass of ganglia that correlates positively with number of neurons infected and rate of reactivation but inversely with number of CD8+ cells present.124,125
Mucosal Immunity
Recent studies mostly involving genital mucosal responses to HSV-2 infection indicate that viral-host interactions in the mucosa dictate clinical expression of disease. A distinct subset of HSV-2 specific CD8+ T cells that persist in genital skin conduct immune surveillance directed at containing virus at the point of release into the epithelium and are first responders capable of controlling HSV-2 and aborting clinical lesions.126 Once virus reaches the dermal-epidermal junction, there are two possible outcomes: subclinical shedding or recurrence defined clinically by a skin blister and ulceration (Fig. 138-2). Histologically, herpetic lesions involve a thin-walled vesicle or ulceration in the basal region, multinucleated cells that may include intranuclear inclusion, necrosis, and an acute inflammatory infection, including neutrophils, natural killer (NK) cells, B cells, and T cells.127 Reepithelialization occurs once viral replication is restricted, almost always in the absence of a scar.