INTRODUCTION
SUMMARY
Thrombophilia refers to laboratory abnormalities that increase the risk of venous thromboembolism (VTE). Over the last several decades numerous factors have been identified. The most prevalent examples of hereditary forms of thrombophilia include the factor V Leiden and prothrombin G20210A mutations; deficiencies of the natural anticoagulants antithrombin, protein C, and protein S; persistently elevated levels of coagulation factor VIII; and mild hyperhomocysteinemia. Taken together, some form of hereditary thrombophilia can be identified in more than 50 percent of patients with VTE who are without obvious reasons for VTE, such as trauma or prolonged stasis. Moreover, hereditary thrombophilia has been associated with arterial cardiovascular disease and obstetric complications such as (recurrent) pregnancy loss and preeclampsia. The high yield of thrombophilia testing has led to widespread testing for these abnormalities in patients. Nevertheless, thrombophilia testing remains a topic of ongoing debate, mostly because of the lack of therapeutic consequences. While hereditary thrombophilia is a clear risk factor for a first VTE, the risk for recurrent episodes is hardly increased compared with nonaffected patients and prolonged anticoagulation is not warranted unless VTE is recurrent. A similar lack of therapeutic consequences applies to patients with arterial cardiovascular disease and women with obstetric complications. Thrombophilia testing in asymptomatic relatives of patients with VTE may be useful in families with antithrombin, protein C, or protein S deficiency, or for siblings of patients who are homozygous for factor V Leiden, and is limited to women who intend to become pregnant or who would like to use oral contraceptives. Careful counseling with knowledge of absolute risks helps patients to making an informed decision in which their own preferences can be taken into account.
Acronyms and Abbreviations
95% CI, 95% confidence interval; APC, activated protein C; ASA, acetylsalicylic acid; HELLP, hemolysis, elevated liver enzymes, low platelets; LMWH, low-molecular-weight heparin; MTHFR, methylenetetrahydrofolate reductase; OR, odds ratio; VKA, vitamin K antagonist; VTE, venous thromboembolism; VWF, von Willebrand factor.
To our knowledge, the term thrombophilia was first used by Nygaard and Brown in 1937, when they described sudden occlusion of large arteries, sometimes with coexistent venous thrombosis.1 In 1956, Jordan and Nandorff extensively reviewed their own and previously published cases on the familial tendency in thromboembolic disease.2 The term thrombophilia was then used to describe patients with prominent manifestations of venous thromboembolism (VTE; venous thrombosis in any site or pulmonary embolism) such as recurrent spontaneous VTE, VTE at young age, a strong family history of VTE, or thrombosis in an unusual site, such as the splanchnic veins or cerebral sinuses. Currently, the term thrombophilia is generally used for laboratory abnormalities, usually in the coagulation system, which increase the risk of VTE. Thrombophilia can be either acquired or hereditary. An example of acquired thrombophilia is the antiphospholipid syndrome, which is characterized by a tendency toward venous or arterial thrombosis or pregnancy complications, in combination with persistent lupus anticoagulant or antibodies to cardiolipin or β2-glycoprotein-1 (Chap. 131). Furthermore, there are many acquired and transient conditions that lead to a prothrombotic state, including cancer, surgery, strict immobilization, pregnancy and the postpartum period, and the use of estrogen-containing medication, such as oral contraceptives and hormone replacement therapy.
Patients with hereditary thrombophilia have an increased risk of developing VTE and just like in patients without thrombophilia, approximately half of patients will develop their first episode in relation to an acquired prothrombotic risk factor. Moreover, despite young age being a criterion for thrombophilia and the mean age at time of a first thrombosis being approximately 10 years lower than in the general population, the majority of patients with thrombophilia will have the first episode later in life.3 The theoretical concept is that patients with thrombophilia have an intrinsic prothrombotic state, which in itself is insufficient to cause thrombosis, but may lead to an event when superimposed upon (clinical) risk factors, including increasing age.3
The role of hereditary thrombophilia in arterial cardiovascular disease has been extensively studied.4,5 Most of those studies did not demonstrate significant associations between hereditary thrombophilia and arterial disease with the exception of patients with events before the age of 55 years.5 Moreover, the relative risk increase was very modest (odds ratios [OR] of 1.1 to 1.8) in studies that did find significant associations, indicating that hereditary thrombophilia is not a major risk factor for arterial cardiovascular disease.4
Like the acquired antiphospholipid syndrome, most hereditary thrombophilias are also modestly associated with pregnancy related disorders such as (recurrent) miscarriage, stillbirth, intrauterine growth retardation, preeclampsia and the hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome of pregnancy, although for later pregnancy complications this association is controversial.6,7,8
In the past decades, hereditary thrombophilia has evolved from a very rare genetic disorder to a prevalent trait. This evolution is a direct consequence of increasing insight into the blood coagulation system, as well as advanced genetic research tools that allowed the search for abnormalities in candidate coagulation proteins and their encoding genes. At present, some form of thrombophilia can be identified in about half of the patients presenting with VTE. Likely inspired by the high yield of thrombophilia testing, testing has increased tremendously for various indications,9 but whether the results of such tests help in the clinical management of patients has still not been settled.10,11 This chapter provides an overview of the most important hereditary thrombophilias and of the history of thrombophilia research. It reviews the risks associated with the most commonly tested thrombophilias and provides guidance on the indications and potential implications of the results of thrombophilia testing in various patient groups.
HISTORY, CLASSIFICATION, PATHOPHYSIOLOGY, AND PREVALENCE OF THROMBOPHILIA
Research into thrombophilia began with the investigation of candidate coagulation proteins and their genes in highly thrombophilic families and linking abnormalities with the clinical phenotype within these families. As a next step, findings were confirmed in case-control studies, which yielded risk increases compared to controls, often derived from the general population. For clinicians and patients however, absolute risk estimates were needed to guide decisions regarding prevention or treatment. These were sought again in family studies of consecutive probands with a specific thrombophilic defect. The major progress in genetic and bioinformatics techniques now allows investigation in populations of patients with VTE, as well as in thrombophilic families.12,13,14
In 1965, deficiency of the natural anticoagulant antithrombin became the first hereditary thrombophilia when Egeberg reported a Norwegian family with a remarkable tendency to VTE.15 Deficiencies of the other anticoagulant proteins, that is, protein C and protein S, were discovered as hereditary risk factors for VTE in the early 1980s.16,17 By that time, genes could be cloned and numerous mutations in the genes encoding antithrombin, protein C, and protein S had been identified as underlying causes of low plasma levels of the anticoagulant proteins.18,19,20 Another decade later, in 1993, Dahlbäck and colleagues described the phenomenon of activated protein C (APC) resistance, a poor anticoagulant response to APC, in a Swedish family with an increased tendency to develop VTE.21 The genetic basis for this APC resistance was discovered independently in several laboratories in 1995 and is caused by a single point mutation in the factor V gene which was termed factor V Leiden.22,23,24,25 In 1996, genetic analysis of prothrombin revealed a G-to-A transition at position 20210 that was more common in patients with VTE and a strong family history of this disease than in healthy controls without VTE.26 In the 1970s, it was found that individuals with non–O blood group have an increased risk of VTE.27 Individuals with non–O blood group have higher levels of von Willebrand factor (VWF) and factor VIII than people with blood group O, which was the presumed mechanism of increased risk. In 1995, data from the Leiden Thrombophilia Study, a case-control study of patients with VTE and matched healthy controls, demonstrated that increased factor VIII (FVIII) activity, but not VWF activity, was independently associated with an increased risk of VTE.28 Homocysteine is an intermediary amino acid formed by the conversion of methionine to cysteine. Homocystinuria or severe hyperhomocysteinemia is a rare autosomal recessive disorder characterized by severe elevations in plasma and urine homocysteine concentrations. This disease is characterized by developmental delay, osteoporosis, ocular abnormalities, and severe occlusive vascular disease. About half of the vascular complications are of venous origin.29 Mild hyperhomocysteinemia was therefore studied as a risk factor for VTE in the 1990s and homocysteine levels exceeding the 95th percentile of the normal population were confirmed to be a risk factor for VTE.30
Since then, numerous genetic variants that increase the risk of VTE to a more or lesser extent have been identified and are variably included in diagnostic panels of thrombophilia testing.31 Essentially, the majority of hereditary thrombophilias exert their effect either by upregulation of procoagulant clotting factors, or by downregulation of anticoagulant factors (Fig. 130–1). An overview of the common hereditary thrombophilias that increase the risk at least twofold, and their prevalence in patients with VTE and in the general population is presented in Table 130–1. For these more common thrombophilias a large number of clinical studies provided reliable estimates of the relative and absolute risk for VTE.
Figure 130–1.
Regulation of blood coagulation. Coagulation is initiated by a tissue factor (TF)–factor VIIa complex that can activate factor IX or factor X. At high TF concentrations, factor X is activated primarily by the TF-VIIa complex, whereas at low TF concentrations, the contribution of the factor IXa–factor VIIIa complex to the activation of factor X becomes more pronounced. Coagulation is maintained through the activation by thrombin of factor XI. The coagulation system is regulated by the protein C pathway. Thrombin activates protein C. Together with protein S, activated protein C (APC) is capable of inactivating factors Va and VIIIa, which results in a downregulation of thrombin generation and consequently in an upregulation of the fibrinolytic system. The activity of thrombin is controlled by the inhibitor antithrombin. The solid arrows indicate activation and the broken arrows inhibition.
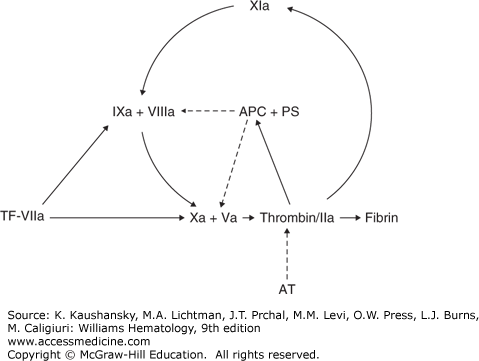
Deficiencies of the natural anticoagulants antithrombin, protein C, and protein S, were among the first established hereditary thrombophilias. For antithrombin and protein C, two types of deficiencies are distinguished. In type I deficiency, levels of both antigen and activity are reduced and in type II, antigen levels are normal, but one or more functional defects in the molecule lead to a decreased activity. Type II antithrombin deficiencies are further subdivided according to the site of the defect in antithrombin. The defect is located in the thrombin binding domain (i.e., the reactive site) in type IIa deficiency, in the heparin binding domain in type IIb deficiency, and type IIc deficiency comprises a pleiotropic group of mutations.32 Interestingly, patients with type IIb deficiency seem to have a significantly lower risk of VTE than other types.32 Protein S circulates in two forms: free protein S (approximately 40 to 50 percent) which functions as a cofactor for APC, and protein S bound to complement component C4b-binding protein. In type I deficiency, total and free antigen levels and activity are all reduced, in type II deficiency, total and free antigen are normal, but activity is reduced, and in type III deficiency, activity and free antigen are reduced, while total antigen is low to normal. Type I and type III are probably phenotypical variants of the same disease as family members with the same DNA mutation can present with either type I or type III deficiency.33 Whether this classification into various subtypes is truly clinically relevant for any of the deficiencies of the natural anticoagulants is largely unknown. Moreover, most laboratory panels now only test the activity of antithrombin, protein C, or protein S, and thereby do not distinguish between different types of deficiencies. Homozygous antithrombin deficiency is extremely rare and the only reported cases involve type IIb deficiencies.34 Homozygous type I deficiency has never been described in humans and is believed to be incompatible with life. Complete antithrombin deficiency in knockout mice leads to embryonic death.35 Homozygous protein C and protein S deficiencies are also very rare and these are associated with neonatal purpura fulminans and massive thrombosis.36,37 In a similar fashion, warfarin-induced skin necrosis has been described in patients with heterozygous protein C or S deficiencies after initiation of vitamin K antagonists (VKAs).38,39 The concept is that after VKA initiation vitamin K–dependent protein C and S levels drop sooner than levels of factors II, IX, and X, thereby temporarily causing a paradoxal procoagulant state.40 This is, however, a rare clinical complication, possibly resulting from concomitant treatment with (low-molecular-weight) heparin in the acute phase of VTE. Deficiencies can be caused by a large number of mutations, that are recorded in occasionally updated databases.18,19,20 Overall, deficiencies of the natural anticoagulants are rare (see Table 130–1). In cohorts of consecutive patients with VTE, the prevalence of a deficiency of one of the natural anticoagulants is below 10 percent.41 In the general population, the prevalence of the deficiencies combined is approximately 1 percent.42,43,44
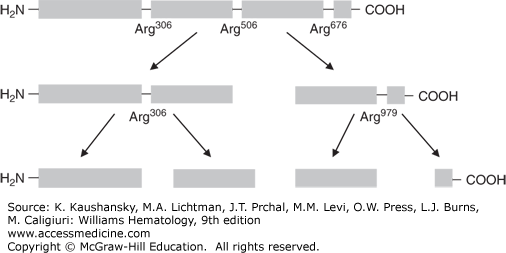
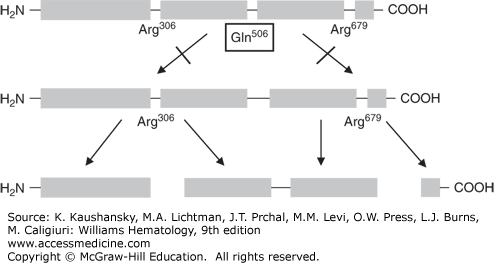
In 1993, Dahlbäck and colleagues first described APC resistance in a Swedish family with a high tendency of VTE.21 In the original paper, Dahlbäck proposed that APC resistance was best explained by an hereditary deficiency of a previously unrecognized cofactor to APC, after having ruled out several possible mechanisms, including deficiencies of protein S and protein C, or linkage with polymorphisms in the FVIII or VWF genes. He then showed that this “cofactor” was identical to coagulation factor V.21 Soon thereafter, several laboratories independently from each other reported the underlying genetic defect, a single G-to-A substitution in the gene of factor V at nucleotide position 1691, resulting in an amino acid change from Arginine (Arg) to Glutamine (Gln) at position 506, the first cleavage site of factor Va for APC22,23,24,25 (Fig. 130–2). The mutation was named factor V Leiden after the city in the Netherlands in which the group with the first publication was located.22 The proteolytic inactivation of activated factor V (FVa) is approximately 10 times slower for Gln506-FVa compared with Arg506-FVa, which explains the partial, but not full, resistance to APC.45 Factor V Leiden is the most common hereditary thrombophilia. In unselected consecutive patients with VTE, the prevalence is 20 to 25 percent.22 The prevalence in the general population varies considerably between different ethnic groups. Factor V Leiden is very rare among Asians and Africans but has a high prevalence (approximately 5 percent) among whites (see Table 130–1).46 Within Europe, the prevalence is higher in the north than in the south.46 This implies a founder effect that suggests that the mutation occurred after the separation of non-Africans from Africans and after the divergence of whites and Asians. Studies using linkage disequilibria between factor V Leiden and specific markers indicate that the mutation occurred around 21,000 years ago.47 The high prevalence of factor V Leiden suggests an evolutionary benefit.48 The presumed mechanism is reduced peripartum and menstrual blood loss in affected female carriers.49,50 More recently, factor V Leiden was associated with increased sperm counts and a shorter conception time in affected male and female carriers, suggesting another evolutionary benefit.51,52,53 The mechanism for this increased fertility is unknown.
Figure 130–2.
Pathophysiology of the factor V Leiden mutation. A. Activated protein C inactivates factor Va by cleaving the protein at the Arginine (Arg)506 cleavage site. B. In carriers of the factor V Leiden mutation, a point mutation in the gene coding for factor V, causes replacement of the amino acid Arginine by Glutamine (Gln) at position 506 of the protein, making factor Va resistant to inactivation by activated protein C (i.e., APC resistance).
The prothrombin G20210A mutation was discovered in 1996 by genetic analysis of the prothrombin gene in families with a strong tendency of VTE and confirmed in a case-control study of patients with VTE.26 The mutation is located in the 3′-untranslated region of the prothrombin gene and augments translation and stability of prothrombin messenger RNA.54 This leads to an average 32 percent higher blood levels of the zymogen prothrombin that is structurally identical to the protein produced by patients with the wild-type gene.26 In unselected patients with VTE, the prevalence of prothrombin G20210A is approximately 6 percent.55 Like factor V Leiden, the prothrombin G20210A mutation is found largely in whites and genetic linkage disequilibrium studies date the mutation back to 24,000 years ago.47,56 Within Europe, the prevalence of the mutation increases from 3 to 5 percent in Southern Europe and the Middle East to 1 to 2 percent in Northern Europe, which opposes the observed geographical gradient of factor V Leiden.56
Most of coagulation FVIII circulates in complex with VWF. Therefore, determinants of VWF, including ABO-blood group and endothelial stimulation, indirectly also determine FVIII levels.57 Furthermore, many other factors have been associated with high FVIII levels, including increasing age, high body mass index, diabetes mellitus, and hypertriglyceridemia.57 High FVIII levels are part of acute phase reactions and sustained increases are observed during pregnancy, surgery, chronic inflammation, malignancy, liver disease, hyperthyroidism, renal disease. In most of these conditions, there is a concordant increase of FVIII and VWF levels.57 Apart from ABO blood group, the genetic causes of high FVIII levels are largely unknown. Nevertheless, persistence over time and familial clustering of high FVIII levels in patients with VTE implies that genetic factors are prevalent. In a family study of consecutive patients with persistently elevated levels of FVIII of at least 150 percent and venous or arterial thrombosis, the prevalence of high FVIII in first degree family members was 40 percent, which is almost in the range to what one would expect from an autosomal dominant inheritance.58 Despite the uncertainty about the mechanisms of high FVIII levels, the association between high FVIII levels and venous thrombosis is well established. The very clear dose-dependent relationship between FVIII levels and the risk of VTE suggests that elevated FVIII is causative for thrombosis.28 Interestingly, increased FVIII levels in patients with VTE are usually not influenced by acute-phase reactions.57 The prevalence of elevated FVIII levels is high: 25 percent of patients with a first episode of deep-vein thrombosis and 11 percent of healthy control subjects have FVIII levels of 150 percent or higher.28 Hence, an increased level of FVIII is the most common familial, albeit not monogenetic, thrombophilia with an estimated population attributable risk of 15 percent.57 Whether measurement of FVIII levels should be part of a hereditary thrombophilia panel is debatable as FVIII levels may be transiently increased by a multitude of external factors including the acute VTE episode itself (Table 130–2). This leads to a significant proportion of “false-positive” test results, a necessity for repeat testing, and potentially unnecessary concern among tested patients.
| Test | Acquired Conditions That Can Cause Abnormal Test Results |
|---|---|
| Increased activated protein C (APC) resistance | Pregnancy, use of oral contraceptives, stroke, presence of lupus anticoagulant, increased factor VIII levels, autoantibodies against APC |
| Factor V Leiden | — |
| Prothrombin G20210A | — |
| Hyperhomocysteinemia | Deficiencies of folate (vitamin B11), vitamin B12, or vitamin B6, old age, renal failure, excessive consumption of coffee, smoking |
| Increased factor VIII levels | Pregnancy, use of oral contraceptives, exercise, stress, older age, acute phase response, liver disease, hyperthyroidism, cancer |
| Decreased level of protein C | Liver disease, use of vitamin K antagonist (VKA), vitamin K deficiency, childhood, disseminated intravascular coagulation, presence of autoantibodies against protein C |
| Decreased level of free protein S | Liver disease, use of VKA, vitamin K deficiency, pregnancy, use of oral contraceptives, nephrotic syndrome, childhood, presence of autoantibodies against protein S, disseminated intravascular coagulation |
| Decreased level of antithrombin | Use of heparin, thrombosis, disseminated intravascular coagulation, liver disease, nephrotic syndrome |
Homocysteine is an intermediate in the metabolism of the amino acids methionine and cysteine and participates in several metabolic pathways. Some of the enzymes involved in homocysteine metabolism are dependent on vitamin B6, folic acid, and vitamin B12, and deficiencies lead to hyperhomocysteinemia. A polymorphism in the enzyme methylenetetrahydrofolate reductase (MTHFR), c.C677T, leads to an alanine to valine substitution at position 222 resulting in a variant enzyme with reduced activity and increased thermolability. Homozygosity for this polymorphism leads to 24 percent increased homocysteine levels and is the most common genetic cause of mild hyperhomocysteinemia.59 The prevalence is 10 to 20 percent in whites and 10 percent in Orientals, but it is rare in Africans.60 Many other conditions are associated with hyperhomocysteinemia, including renal failure, hypothyroidism, smoking, excessive coffee consumption, inflammatory bowel disease, psoriasis, and rheumatoid arthritis.61 Severe hyperhomocysteinemia (plasma levels exceeding 100 μmol/L), also named homocystinuria, is a rare autosomal recessive disorder clearly associated with vascular occlusive disease.29 Because of this association, mildly increased levels (exceeding the 95th percentile of the normal population) were studied as a risk factor for venous and arterial thrombosis. In the Leiden Thrombophilia Study mild hyperhomocysteinemia was associated with a 2.5-fold increased risk of first VTE.30 Interestingly, the association of homocysteine levels with VTE was stronger in men than in women and increased with age.30 The mechanism by which hyperhomocysteinemia would lead to thrombosis is unknown. A 2005 meta-analysis that included more than 50 studies with more than 8000 cases of VTE confirmed the association of VTE with hyperhomocysteinemia and also demonstrated a 20 percent higher VTE risk in MTHFR 677TT carriers compared with 677CC carriers.62 The association between homocysteine levels is again under scrutiny in newer studies. The single largest case-control study of more than 4000 patients with a first VTE found no association between MTHFR 677TT and VTE.63 Furthermore, a family study of probands with mild hyperhomocysteinemia and venous or arterial thrombosis found that the association between homocysteine and VTE disappeared after adjustment for FVIII levels.64 Finally, homocysteine lowering by B vitamins did not reduce the incidence of recurrent events both in patients with VTE as well as in patients with arterial cardiovascular disease.65,66 As a result of these studies, homocysteine testing (either its levels of polymorphisms) as part of a thrombophilia panel has been largely abandoned.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


