INTRODUCTION
SUMMARY
Abnormalities of platelet function manifest themselves primarily as excessive hemorrhage at mucocutaneous sites, with ecchymoses, petechiae, epistaxis, gingival hemorrhage, and menorrhagia most common. Both quantitative and qualitative platelet abnormalities can produce these symptoms, so it is necessary to exclude thrombocytopenia (Chap. 117) by performing a platelet count. Chapter 121 discusses acquired qualitative platelet abnormalities and this chapter discusses the hereditary qualitative platelet abnormalities.
The hereditary qualitative platelet disorders can be classified according to the major locus of the defect (see Table 120–1 and Fig. 120–1). Thus, abnormalities of platelet glycoproteins, platelet granules, and signal transduction and secretion can all result in hemorrhagic diatheses and prolonged bleeding times. Glanzmann thrombasthenia results from abnormalities in one of two integrin subunits, either αIIb (glycoprotein [GP] IIb) or β3 (GPIIIa), resulting in loss or dysfunction of the αIIb β3 (GPIIb/IIIa) receptor. This results in a profound defect in platelet aggregation and secondary defects in platelet adhesion, secretion, and platelet coagulant activity. Heterozygous gain of function mutations in αIIbβ3 can result in a syndrome of macrothrombocytopenia. Loss of the platelet GPIb–IX–V complex because of abnormalities in GPIbα, GPIbβ, or GPIX results in the Bernard-Soulier syndrome, which is characterized by giant platelets and modest thrombocytopenia. The major defect is in platelet adhesion because of a decrease in platelet interactions with von Willebrand factor, but abnormalities in αIIbβ3 activation and thrombin-induced aggregation are also present. A gain of function defect in GPIbα (platelet-type [pseudo-] von Willebrand disease) can produce a hemorrhagic disorder via depletion of high-molecular-weight von Willebrand multimers. Inherited defects in platelet dense or α granules, agonist receptors, or proteins and mechanisms involved in signal transduction and secretion also lead to platelet dysfunction and produce hemorrhagic symptoms.
Abnormalities of platelet coagulant activity, that is, the ability of platelets to facilitate thrombin generation (Chap. 112), can lead to a hemorrhagic diathesis. Impaired platelet function may occur in association with mutations in transcription factors RUNX1, GATA-1, FLI-1, and GFI1B, and these patients have thrombocytopenia as well.
Acronyms and Abbreviations
ADP, adenosine diphosphate; BSS, Bernard-Soulier syndrome; βTG, β-thromboglobulin; BLOC, biogenesis of lysosome-related organelles complex; cAMP, cyclic adenosine monophosphate; EDTA, ethylenediaminetetraacetic acid; GFI1b, growth factor independent 1B; GPS, gray platelet syndrome; GT, Glanzmann thrombasthenia; HLA, human leukocyte antigen; HPS, Hermansky-Pudlak syndrome; Ig, immunoglobulin; LAD, leukocyte adhesion deficiency; MIDAS, metal ion-dependent adhesion site; PAR, protease-activated receptor; PF4, platelet factor 4; PKC; protein kinase C; PLC, phospholipase C; rFVIIa, recombinant factor VIIa; TGF, transforming growth factor; TXA2, thromboxane A2; VWD, von Willebrand disease; VWF, von Willebrand factor.
PLATELET FUNCTION IN HEMOSTASIS
Abnormalities of platelet function manifest themselves primarily as excessive hemorrhage at mucocutaneous sites, with ecchymoses, petechiae, epistaxis, gingival hemorrhage, and menorrhagia being most common. Mild platelet function abnormalities will not cause spontaneous bleeding but may cause (excessive) hemorrhage after trauma or medical interventions. Both quantitative and qualitative platelet abnormalities can produce these symptoms, so it is necessary to exclude thrombocytopenia (Chap. 117) by performing a platelet count. Although no longer performed widely, a prolonged bleeding time in a patient with a normal platelet count is suggestive of a qualitative platelet abnormality. Some patients may have abnormalities in both platelet number and function. Chapter 121 discusses acquired qualitative platelet abnormalities and this chapter discusses hereditary qualitative platelet abnormalities.
Following injury to the blood vessel, platelets adhere to exposed subendothelium by a process that involves, among other events, the interaction of a plasma protein, von Willebrand factor (VWF), and a specific glycoprotein complex on the platelet surface, the glycoprotein (GP) Ib–IX–V complex. Adhesion is followed by recruitment of additional platelets that form clumps (aggregation), which involves binding of fibrinogen to specific platelet surface receptors, a complex comprised of integrin αIIbβ3 (GPIIb-IIIa). Platelet activation is required for fibrinogen binding; resting platelets do not bind fibrinogen. Activated platelets release the contents of their granules (secretion), including adenosine diphosphate (ADP) and serotonin from the dense granules, which causes the recruitment of additional platelets. Moreover, platelets play a major role in coagulation mechanisms; several key enzymatic reactions occur on the platelet membrane phospholipid surface. A number of physiologic agonists interact with platelet surface receptors to induce responses, including a change in platelet shape from discoid to spherical (shape change), aggregation, secretion, and thromboxane A2 (TXA2) production. The binding of agonists to their platelet receptors initiates numerous intracellular events (Chap. 112) including the production or release of several messenger molecules. One pathway leads to the hydrolysis of phosphoinositide (PI) by phospholipase C leading to the formation of diacylglycerol and inositol 1,4,5-triphosphate [IP3]). These and other mediators induce or modulate the various platelet responses of Ca2+ mobilization, protein phosphorylation, aggregation, secretion, and thromboxane production. Numerous other mechanisms, such as activation of tyrosine kinases and phosphatases, are also triggered by platelet activation (Chap. 112). Inherited or acquired defects in the above and other platelet mechanisms may lead to impaired platelet function and a bleeding diathesis.
CLASSIFICATION OF HEREDITARY QUALITATIVE PLATELET DISORDERS
The hereditary qualitative platelet disorders can be classified according to the major locus of the defect (Table 120–1 and Fig. 120–1). Glanzmann thrombasthenia (GT) is caused by abnormalities in either integrin αIIb (GPIIb) or β3 (GPIIIa), resulting in loss or dysfunction of the integrin αIIbβ3 receptor. This results in a profound defect in platelet aggregation and secondary defects in platelet adhesion, secretion, and coagulant activity. Loss of the platelet GPIb–IX–V complex because of abnormalities in GPIbα, GPIbβ, or GPIX results in the Bernard-Soulier syndrome (BSS), which is characterized by giant platelets and thrombocytopenia. The major defect is in platelet adhesion owing to a decrease in platelet interactions with VWF. A gain-of-function defect in GPIbα (platelet-type [pseudo] von Willebrand disease [VWD]) can also produce a hemorrhagic disorder via depletion of high-molecular-weight VWF multimers. Defects in secretion of granule contents because of deficiencies in the granules or in the mechanisms that mediate secretion results in impaired platelet function. Inherited defects in agonist receptors or proteins or mediators involved in signal transduction or thromboxane synthesis may also produce hemorrhagic symptoms. Abnormalities of platelet coagulant activity, that is, the ability of platelets to facilitate thrombin generation (Chap. 113), and in cytoskeletal-linking proteins, can also lead to a hemorrhagic diathesis. Lastly, it is becoming clear that some patients may have abnormalities multiple aspects of platelet function and number related to a mutation in a hematopoietic transcription factor that regulates gene expression in megakaryocytes and platelets.
Figure 120–1.
Evaluation of patients for inherited abnormalities in platelet number or function. The major and well-recognized entities are shown here. A reduced platelet count occurs in patients with purely quantitative platelet disorders (inherited or acquired) as well as in patients who have inherited qualitative platelet disorders. Chapter 117 discusses inherited thrombocytopenias. Notable among patients with thrombocytopenia and inherited platelet dysfunction is the Bernard-Soulier syndrome (BSS), which is characterized by giant platelets. Patients with the Gray platelet syndrome are characterized by thrombocytopenia and gray appearance of platelets on the blood smear due to paucity of granules. Platelet aggregation studies can provide clues regarding the nature of the underlying platelet abnormality. Decreased response to ristocetin alone with normal responses to other agonists is found in the BSS and von Willebrand disease (VWD; type I and type III). The response to ristocetin is enhanced in VWD type IIB and the platelet-type VWD. Impaired response to collagen or epinephrine alone may suggest a defect in their respective receptors. Patients with adenosine diphosphate (ADP) and thromboxane receptor defects have impaired responses to multiple agonists because of the feedback amplification provided by ADP and thromboxane A2 (TXA2) when activated by different agonists. Absence of both primary and secondary waves of aggregation in response to all physiologic agonists occurs in Glanzmann thrombasthenia (GT). A heterogeneous group of platelet defects are characterized by a decreased secondary wave of platelet aggregation in response to ADP and epinephrine, and diminished responses to low doses of collagen, TXA2 and thrombin. They can be broadly separated into granule defects (involving dense [δ] or both dense and α granules) and defects in the platelet secretion or release reaction associated with normal dense granule stores. The granule defects may occur in isolation or in association with other syndromes. Secretion abnormalities arise from defects in mechanisms that regulate the release of granule contents, and include defects at the level of platelet receptors (ADP, TXA2), signaling events involving guanosine triphosphate (GTP)–binding proteins that link surface receptors to intracellular enzymes, phospholipase C activation, and protein phosphorylation (protein kinase C [PKC]-θ). They may also arise from defects in TXA2 synthesis because of deficiencies of phospholipase A2 (PLA2), cyclooxygenase, or thromboxane synthase. Patients with the Scott syndrome are characterized by a normal bleeding time, normal responses in aggregation studies, and a shortened prothrombin time, which reflects the defect in the platelet–coagulant protein interactions. Additional details on the various entities are described in the section “General Approach to Patients with Mucocutaneous Bleeding Symptoms for Abnormalities in Platelet Number or Function.” GP, glycoprotein; PLA2, phospholipase A2; PLC, phospholipase C; SPD, storage pool deficiency; u-PA, urokinase plasminogen activator; VWF, von Willebrand factor.

|
CLINICAL MANIFESTATIONS
Disorders of platelet function are characterized by highly variable mucocutaneous bleeding manifestations and excessive hemorrhage following surgical procedures or trauma. These include ecchymoses, petechiae, epistaxis, gingival bleeding, and menorrhagia. Spontaneous hemarthrosis are distinctly rare, distinguishing them from the hemophilias, and deep hematomas and spontaneous central nervous system bleeding are highly unusual in these patients. Postpartum hemorrhage and postsurgical bleeding may be severe in some patients. In general, most patients with platelet function defects have mild to moderate bleeding manifestations, but may be severe in some entities, such as the GT and the BSS. Individual patients with the same functional defect or even the same genetic defect may also vary in the intensity of bleeding manifestations, suggesting one or more disease-modifying genes exist. Moreover, the severity of bleeding symptoms may vary over the lifetime of the same individual, implying that factors in addition to the platelet defect may be contributing to the bleeding risk. Sometimes, a mild platelet function defect becomes clinically manifest when the patient uses medication interfering with primary hemostasis (e.g., nonsteroidal antiinflammatory drugs [NSAIDs]).
Although normal in many of the inherited platelet function defects, the platelet count may be decreased in some entities, such as the BSS and the gray platelet syndrome (GPS) and in association with mutations in hematopoietic transcription factors. Most patients with platelet function defects, but not all, have a prolonged bleeding time, a test no longer available in most centers because of its inherent inaccuracies. In vitro platelet aggregation and secretion studies provide evidence for the dysfunction but are not generally predictive of the severity of clinical manifestations. In some patients, such as those with abnormal platelet coagulant activities, these studies may be normal. Some patients with platelet dysfunction are initially detected through abnormalities on testing with the platelet function analyzer (PFA-100).
GENERAL APPROACH TO PATIENTS WITH MUCOCUTANEOUS BLEEDING SYMPTOMS FOR ABNORMALITIES IN PLATELET NUMBER OR FUNCTION
Platelet disorders are characterized by alterations in platelet number or function or both. A general approach is shown in Fig. 120–1. A reduced platelet count can occur as an isolated platelet disorder (inherited or acquired) or with evidence of a concomitant defect in platelet function. Platelet size and examination of the blood film may provide insights into the underlying mechanism or cause. Platelet size provides clues in some entities (Chap. 117).1,2 Decreased platelet size is characteristic of the Wiskott-Aldrich syndrome. In the Paris-Trousseau/Jacobsen syndrome, thrombocytopenia is associated with giant α granules in a subpopulation of platelets in association with mutations in the transcription factor FLI1. Transcription factor RUNX1 mutations are associated with familial thrombocytopenia, abnormal platelet function, and predisposition to leukemia. Large platelets that lack the purple granules on the peripheral smear are observed in the GPS (α-storage pool disease). The diagnosis is obtained with biochemical analysis of α-granule contents. Patients with platelet-type (pseudo-) VWD and type 2b VWD have moderate thrombocytopenia and large platelets. Studies of GPIb function and biochemistry establish the diagnosis. Patients who are hemizygous for GPIbβ because of deletion of 22q11.2, those with mutations in transcription factor GATA-1 or β1 tubulin (R318W), and some patients who are heterozygous for defects in GPIb/IX have variable thrombocytopenia and large platelets. Mutations that activate αIIbβ3 are also associated with large platelets and thrombocytopenia. The platelets in BSS are truly giant; the diagnosis is confirmed with biochemical and functional analyses of the GPIb–IX–V complex.
A variety of methods have been developed to assess platelet function and new instrumentation continues to be developed.3,4,5,6,7,8 Platelet aggregation studies performed using platelet-rich plasma can loosely separate patients into those with defects in the primary wave of platelet aggregation (dependent on either fibrinogen, VWF, their respective receptors, or agonist receptors for collagen, ADP, TXA2) and those with defects in the secondary wave of aggregation. Enhanced ristocetin-induced platelet aggregation at low doses of ristocetin is characteristic of patients with platelet-type VWD (who have a defect in the GPIb receptor) and patients with type 2b VWD (who have gain-of-function defect in VWF) (Chap. 126). These two diseases differ in the binding of the patient’s VWF to normal platelets, or the ability of purified VWF, cryoprecipitate or asialo-VWF to aggregate patient platelets; the diagnosis of platelet-type VWD or its confirmation requires genetic analysis of GPIb.
Neither ristocetin nor the snake venom botrocetin induces platelet aggregation if the plasma lacks functional VWF, as in VWD (Chap. 126), or if the platelets lack functional GPIb–IX complexes, as in BSS. The defect in VWD, but not BSS, can be corrected by adding normal plasma or purified VWF. Direct analysis of VWF and the platelet GPIb–IX complex are used to confirm the diagnosis.
Patients whose plasma lacks fibrinogen (afibrinogenemia; Chap. 125) or whose platelets cannot bind fibrinogen because of abnormal αIIbβ3 receptors (GT) or inability to activate integrin αIIbβ3 (leukocyte adhesion deficiency [LAD]-3) as the result of a kindlin-3 abnormality will have no primary wave of platelet aggregation in response to all physiologic agonists, including ADP, epinephrine, collagen, TXA2, and thrombin. Simple coagulation tests (prothrombin time, partial thromboplastin time, and measurement of plasma fibrinogen) and analysis of platelet integrin αIIbβ3 receptors, and kindlin-3 can differentiate between these two groups. Isolated defects in the primary response to collagen have been observed in patients with abnormalities in platelet integrin α2β1 (GPIa/IIa) or GPVI. Platelet glycoprotein analysis can separate these from each other. Because antibodies to GPVI can result in receptor depletion from circulating platelets, a search for anti-GPVI should be undertaken in patients with reduced platelet GPVI. Defects in ADP, epinephrine, or TXA2 receptors will result in decreased platelet aggregation in response to the specific agonist. However, patients with isolated ADP and TXA2 receptor abnormalities have impaired aggregation in response to other agonists as well because of the feedback potentiation provided by ADP and TXA2.
A very heterogeneous group of platelet defects can result in a decreased secondary wave of platelet aggregation in response to ADP and epinephrine and diminished responses to low doses of collagen and thrombin. They can be separated into granule defects and defects in platelet secretion or the release reaction. Operationally, these two groups can be separated on the basis of their release of dense granule contents in response to high doses of thrombin. High-dose thrombin activation can overcome most or all of the release reaction (secretion) abnormalities, so platelets from patients with these disorders will release normal amounts of granule contents; in contrast, patients with reduced granule contents have abnormal granule release responses even when using high doses of thrombin. α-Granule contents and dense-body contents can be measured immunologically and biochemically; electron microscopy can establish granule defects. Specific analysis of the genes or proteins implicated in the different granule biogenesis abnormalities (Wiskott-Aldrich syndrome [WASP], Hermansky-Pudlak syndrome [HPS1–9], Chédiak-Higashi syndrome [LYST], Paris-Trousseau/Jacobson syndrome [FLI1], and inherited platelet disorder with predisposition to leukemia [RUNX1]) can establish the diagnosis. The Quebec platelet disorder is characterized by increased urokinase plasminogen activator (u-PA) in α-granules and degradation of several α-granule proteins. The diagnosis can be established by immunoblot analysis or analysis of u-PA activity and confirmed by genetic analysis of u-PA. Secretion abnormalities arise as a result of defects in mechanisms that regulate the secretion of granule contents, and may include abnormalities at various levels, including surface receptors, guanosine triphosphate (GTP)-binding proteins that link surface receptors to intracellular enzymes, phospholipase C (PLC) activation, and protein phosphorylation (protein kinase C [PKC]-θ). They also arise from defects in TXA2 synthesis caused by deficiencies of phospholipase A2 (PLA2), cyclooxygenase, or thromboxane synthase. Specific studies on signal transduction mechanisms, PI metabolism, Ca2+ mobilization, protein phosphorylation, and thromboxane production are needed to define these defects. Because transcription factor abnormalities can affect the expression of multiple proteins involved in megakaryopoiesis and platelet function, they can simultaneously produce alterations in platelet count, structure, and function.
In the disorder of platelet coagulant activity (Scott syndrome) platelet aggregation studies are normal and the serum prothrombin time is the preferred screening assay. Other tests of platelet coagulant activity, microvesiculation, and phospholipid transfer are used to establish the diagnosis.
The introduction of microfluidic multiparameter assessments of platelet function,7,8 coupled with advances in proteomics, RNA expression profiling, and DNA sequencing are shifting the diagnosis of platelet function disorders from a target gene approach to one in which unbiased comprehensive functional and genetic analyses are employed. These methods have identified mutations in RUNX1 and FLI-19 in patients with platelet function disorders, in NBEAL2 in GPS,10,11,12 in TMEM16 in the Scott syndrome,13 and in RBM8A in thrombocytopenia with absent radii (TAR) syndrome.14,15 Many additional genetic alterations, including ones that affect multiple systems, are likely to be identified in the near future as these techniques are employed more broadly.
ABNORMALITIES OF ADHESION RECEPTORS
GT is an inherited hemorrhagic disorder characterized by a severe reduction in, or absence of, platelet aggregation in response to multiple physiologic agonists as a result of qualitative or quantitative abnormalities of platelet integrin αIIb (GPIIb; CD41) and/or integrin β3 (GPIIIa; CD61).16
In 1918, Eduard Glanzmann, a Swiss pediatrician, described a group of patients with hemorrhagic symptoms and a defect in platelet function, namely the ability to retract clots (“weak” platelets or thrombasthenia).17 Subsequent studies demonstrated that thrombasthenic patients have prolonged bleeding times and that their platelets fail to aggregate in response to physiologic agonists18,19,20,21 and have markedly reduced18,20,21,22 platelet fibrinogen. In the mid-1970s, Nurden and Caen23 and Phillips and colleagues24 discovered that thrombasthenic platelets are deficient in both integrin αIIb and β3. Later studies demonstrated that integrin αIIb and β3 form a calcium-dependent complex in the platelet membrane that functions as a receptor for fibrinogen and other adhesive glycoproteins.25,26,27,28 Cloning and sequencing of the complementary DNAs for integrin αIIb29 and β330 identified them as separate protein subunits that are members of the integrin receptor superfamily31 and permitted the molecular biological characterization of patients with the disorder (see database of Glanzmann patients http://med.mssm.edu/glanzmanndb).
GT is a rare disorder characterized by autosomal recessive inheritance with a worldwide distribution. In regions where consanguineous matings are common, groups of patients with the disorder have been identified, and in several populations founder mutations have been identified by analyzing polymorphisms in the DNA surrounding the affected mutation. These include 42 patients from South India; 39 patients from the Iraqi-Jewish population in Israel; 46 Arab patients from Israel, Jordan, and Saudi Arabia; 30 patients from Italy; a smaller number of patients from three Gypsy families; and 43 patients from Pakistan.22,32,33,34,35,36,37,38,39,40 Perhaps the highest frequency of a GT mutation is found in the Iraqi-Jewish population where the most common mutation causing GT was found in six of 700 individuals.39
The platelet integrin αIIbβ3 receptor is required for platelet aggregation induced by all physiologic agonists (ADP, epinephrine, thrombin, collagen, TXA2) (Chap. 112).41 Consequently, abnormalities in the receptor result in a failure of platelet plug formation at sites of vascular injury, and excessive bleeding.
The integrin αIIbβ3 receptor is also responsible for the uptake of fibrinogen from plasma into α granules,42 hence, patients with GT have markedly reduced platelet fibrinogen.18,20,21,43,44 Clot retraction requires platelets with intact integrin αIIbβ3 receptors,45,46 and is, therefore, usually abnormal in GT.18
Defects in either integrin αIIb or β3 result in the same functional defect because both subunits are required for receptor function (Chap. 112). Biosynthetic studies indicate that integrin αIIb and β3 form a complex soon after protein synthesis in the rough endoplasmic reticulum47,48,49; subsequent posttranslational processing50 and transport to the platelet membrane require that the complex be intact (Fig. 120–2).51,52 Complex formation protects each of the glycoproteins from proteolytic digestion,47,48,49,50 so if either integrin αIIb or β3 is absent or unable to form a normal complex, the other subunit will be rapidly degraded, most likely through a proteasomal mechanism. Thus, a deficiency in either glycoprotein produces a deficiency in both. Because complex formation and vesicular transport are also required for proteolytic processing of pro-αIIb into its constituent αIIbα and αIIbβ subunits,50 if these processes do not occur normally, the very small amount of residual integrin αIIb will be pro-αIIb, not mature αIIb.53 Pro-αIIb has been reported to bind to the membrane-bound endoplasmic reticulum chaperone calnexin, providing a potential mechanism for assessing whether the protein has undergone proper folding (calnexin cycle) and perhaps explaining how the receptor adopts a bent configuration.54,55
Figure 120–2.
Biogenesis of integrin αIIbβ3 and integrin αVβ3 receptors. The nuclear genes for integrins αIIb (chromosome localization 17q21.32; gene designation ITGA2B; 30 exons), αV (2q31; ITGAV; 30 exons), and β3 (17q21.32; ITGB3;14 exons) are transcribed into messenger RNA and translated by ribosomes attached to the membranes of the endoplasmic reticulum (ER). The proteins undergo initial glycosylation and form the integrins αIIbβ3 and αVβ3 heterodimers in the ER. It is presumed that many more integrin αIIbβ3 complexes form than αVβ3 complexes because the final copy number of platelet integrin αIIbβ3 receptors is approximately 100,000 whereas it is only 50 to 100 for αVβ3. This is shown schematically by the differences in the width of the arrows depicting integrin αIIbβ3 versus αVβ3 complex formation. The heteroduplexes are transported to the Golgi where the carbohydrate chains undergo modification to their mature structures and both αIIb and αV subunits undergo proteolytic cleavage within a disulfide-bonded loop, resulting in two-chain forms of the receptor subunits. Mature integrin αIIbβ3 receptors are transported to α-granule membranes, where they undergo cycling to and from the plasma membrane. This process results in the internalization of fibrinogen and perhaps other plasma proteins. Integrin αIIbβ3 may be transported directly to the plasma membrane and is transported to α granule membranes. Of the total of approximately 100,000 integrin αIIbβ3 receptors, approximately two-thirds are on the surface at any given time and the remaining one-third can be brought to the surface by platelet activation. The distribution of integrin αVβ3 between the plasma membrane and α granules, and the potential cycling of the receptors between α granules and the plasma membrane have not be defined. (Used with permission from Dr. W. Beau Mitchell, New York Blood Center, New York, NY.)
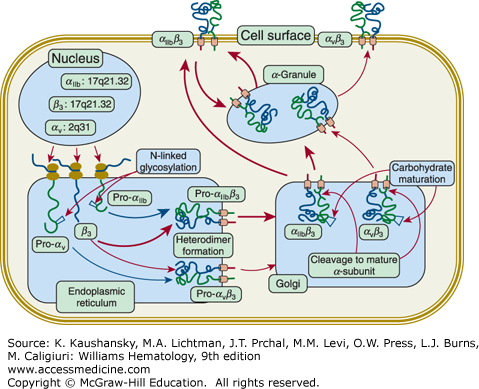
Integrin β3 (GPIIIa) can also combine with the integrin αV (CD51) subunit to form the integrin αVβ3 “vitronectin” receptor30,56,57 (see Fig. 120–2; Chap. 112). This receptor can bind many of the same adhesive glycoproteins as integrin αIIbβ3, although there are some differences in ligand preference and binding sequences.57,58,59,60,61 A small number of integrin αVβ3 receptors are present on platelets (50 to 100 per platelet)60,62,63; osteoclasts, endothelial cells, macrophages, vascular smooth muscle, and uterine cells, among others, also have integrin αVβ3 receptors.64,65 In general, GT patients with defects in integrin β3 also are deficient in integrin αVβ3, whereas patients with defects in integrin αIIb have either normal or increased numbers of platelet integrin αVβ3 receptors.60,63,64,66,67,68 One exception to this rule is a patient with a defect in β3 (H280P) that interferes with integrin αIIbβ3 biogenesis to a much greater extent than integrin αVβ3 biogenesis.69 At present, there is no evidence that patients who lack integrin αVβ3 receptors in addition to lacking integrin αIIbβ3 receptors have a more-severe hemorrhagic diathesis or suffer from any other abnormalities, perhaps because alternative receptors containing integrin αV associated with other β subunits can substitute for integrin αVβ3.63 Upregulation of integrin α2β1 on osteoclasts of Iraqi-Jewish patients with GT has been reported as a potential compensatory mechanism to explain the lack of bone changes despite the deficiency in osteoclast integrin αVβ3.70
The molecular biologic abnormalities in more than 100 patients with GT have been identified and they are listed in an internet database that is updated continuously71 (http://med.mssm.edu/glanzmanndb). Figure 120–3 contains information on mutations of particular interest. Of note, many of the patients with identified mutations are compound heterozygotes rather than homozygotes, indicating that a sizable number of silent carriers are present in the population. Where consanguinity is common, the disorder is more likely to be caused by a homozygous mutation arising in a founder, but even under these circumstances, more than one mutation may be present. Thus, in the Iraqi-Jewish population, in which consanguinity has been present from 586 bce to the present, two separate mutations have been identified in more than one family.39 Most of the missense mutations result in decreased expression of integrin αIIbβ3 on the surface of platelets. This probably reflects the stringent structural requirements for proper folding and complex formation.
Figure 120–3.
Diagram of αIIbβ3 structure and identification of select mutations causing Glanzmann thrombasthenia. The website http://med.mssm.edu/glanzmanndb contains a full listing of reported Glanzmann thrombasthenia mutations. The αIIbβ3 structure depicted is a composite of data from crystal and nmr structures, as well as molecular modeling of missing regions. Among the missense mutations identified are ones that (1) interfere with inside-out and outside-in signaling (β3 S752P); (2) interfere with ligand binding to either the metal ion-dependent adhesion site (MIDAS) in β3 (β3 D119Y and D119N) or the αIIb component of the ligand binding site (Y143H, P145L/A, insert R160/T161); (3) result in receptors that are sensitive to dissociation by divalent cation chelation (β3 R214W, R214Q, R216Q); (4) result in a constitutively active receptor (β3 C560R); (5) alter the interface between αIIb and β3 and disrupt ligand binding (β3 L262Y); (6) result in a β3 protein that can complex more effectively with αV than αIIb (S162L, R216Q, H280P); or (7) alter the αIIb propeller structure and prevent normal αIIbβ3 complex formation, processing, and/or transport. The mutations identified by number 8 in αIIb (G991C and R995Q/W) and β3 (L718P and D723H) are gain-of-function mutations associated with macro/anisothrombocytopenia. (Reproduced with permission from Dr. Ana Negri based on PDBids 3FCS, 3G9W, 2K9J, 2KNC, and 2KV9 and molecular modeling of the missing segments of the αIIb calf domain, the β3 hybrid domain, and the link between the β3 EGF-1 and EGF-2 domains.)
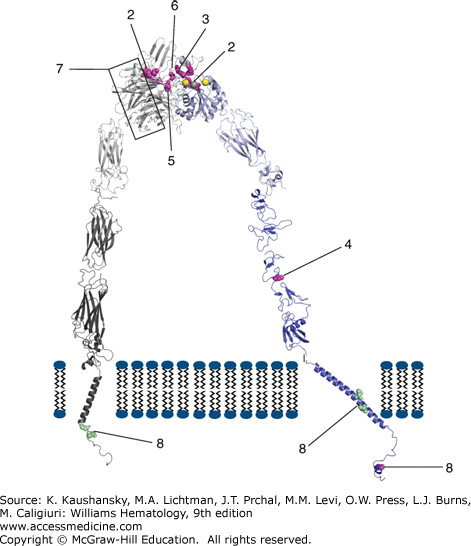
Mutations in Integrin αIIbβ3 Within the Metal Ion-Dependent Adhesion Site of Integrin β3 and the Interface with the Integrin αIIb β-Propeller A metal coordination site or MIDAS domain, which is highly conserved in six integrin receptor α-chain subunits and required for ligand-binding, is also present in the β-A (or I-like) domain of the integrin β3 subunit.72 Mutagenesis and molecular modeling experiments suggested that a highly conserved DxSxS amino acid sequence73 motif plus additional coordinating residues are brought together in the three-dimensional structure of the β3 subunit to form a cation-binding sphere of the MIDAS domain,74 and this was confirmed by the crystal structures of integrin αVβ3 and later integrin αIIbβ3 (see Chap. 112, Fig. 112–111, and Fig. 120–3).75,76 Thus, the β3 MIDAS is composed of Asp119, Ser121, Ser123, Glu220, and Asp251. A region originally termed the ligand-associated metal binding site (LIMBS) in integrin αVβ3,77 but now termed the synergy metal binding site (SyMBS) in integrin αIIbβ3,78 binds a Ca2+ ion and is required for binding of ligands to the MIDAS. It is composed of atoms from D158, N215, D217, P219, and E220. Integrin β3 residues 214 and 216 are in close proximity with both the SyMBS residues and the interface with the αIIb subunit. Adjacent to the MIDAS domain is a metal ion site termed the ADMIDAS (adjacent to metal ion-dependent adhesion site), in which calcium is coordinated by Ser123, Asp126, Asp127, and Met335 in unliganded integrin αVβ3 and integrin αIIbβ3, but Asp251 substitutes for Met335 in the ligand-bound structures of both integrin αVβ3 and integrin αIIbβ3. The crystal structures also demonstrated that peptide ligands containing the Arg-Gly-Asp (RGD) cell adhesion sequence interact with integrin αIIbβ3 and integrin αVβ3 in part by coordination of the metal ion in the MIDAS by the aspartic acid in the RGD peptide.77,79 The low-molecular-weight drugs eptifibatide and tirofiban, which block ligand binding to the αIIb subunit, have negatively charged regions that also interact with the MIDAS cation.76 The fibrinogen γ-chain C-terminal dodecapeptide mediates binding to integrin αIIbβ3 and a crystal structure of the complex demonstrates that an aspartic acid carboxyl oxygen coordinates the MIDAS cation whereas the carboxy terminal valine interacts with the nearby cation in the ADMIDAS.76,79 A number of mutations in patients with GT have been identified within the cation-binding sphere of the MIDAS domain (see Fig. 120–3, and Chap. 112, Fig. 112–11). Two mutations D119Y (Cam variant)80 and D119N (patient NR),81 are located within the conserved DxSxS amino acid motif and produce severe abnormalities of ligand binding to integrin αIIbβ3, but do not affect its surface expression. Mutations at residues R214 and R216 result in abnormal integrin αIIbβ3 receptors that cannot bind ligand and are very sensitive to dissociation by calcium chelation, perhaps because they are at the integrin αIIb–β3 interface.32,82,83,84 Disrupting the SyMBS with a D217V mutation also leads to GT despite the expression of normal amounts of the integrin protein.85 Further support for the importance of the MIDAS domain, SyMBS, and adjacent residues comes from studies in which the mutations D119N, R214W, D217N, E220Q, and E220K were introduced into Chinese hamster ovary CHO cells in vitro and shown to result in functional abnormalities.86
The interface between the αIIb β-propeller and the β3 subunit also involves, in part, the interaction between β3 R261, contained in a four-amino-acid 310 helix, with a number of hydrophobic residues in the αIIb β-propeller arranged as inner and outer rings, making up a cage.75 A β3 subunit L262Y mutation, adjacent to R261 results in disruption of the helix and an unstable integrin αIIbβ3 complex that is expressed on the surface of platelets but is unable to bind fibrinogen.87 The platelets of the patient with this mutation were able to bind fibrin and support clot retraction, suggesting different requirements for fibrinogen and fibrin binding.
Mutations in Integrin αIIbβ3 Within the αIIb β–Propeller Sequence Based on their homology to another integrin α subunit, the aminoterminal 450 amino acids of integrin αIIb and the homologous region in integrin αV, which contain the minimal ligand-binding sequence,88 were predicted to fold into seven repeat (blade) β-propellers, containing four cation-binding sites,89 and this prediction was confirmed by the crystal structures of both αV and αIIb integrin subunits.75,76 The upper surface of the propeller interacts with the β3 subunit β-A (or I-like) domain to form the head of the integrin αIIbβ3 complex, which is the site of ligand binding. Each repeat (blade) contains four β strands that are connected by loops. The four calcium-binding sites in the αIIb subunit, which are in β-hairpin structures, are located in loops on the undersurface of the propeller. Ligand binding in integrin αIIb has been localized to a hydrophobic (F160, Y190, F231) and negatively charged (D224) pocket that lies adjacent to the MIDAS domain in the β3 subunit, and is composed of contributions from the loops that link blade 2 to blade 3 (residues 144 to 171), β strand 2 to β strand 3 in blade 3 (residues 186 to 193), and blade 3 to blade 4 (residues 223 to 236). Integrin αIIb contains a unique “cap” subdomain made up of four insertions in β propeller loops (residues 72 to 88, 111 to 126, 147 to 166, 200 to 217) that also plays a role in ligand binding.76
GT missense mutations located within the integrin αIIb β-propeller (see Fig. 120–3) primarily affect transport of the integrin αIIbβ3 complex to the cell surface,68,90,91,92,93 but several missense mutations and an insertion result in functionally defective receptors. Thus, Y143H affects soluble ligand binding but not adhesion or clot retraction,94 and P145A, which has been identified in several kindreds,32,95 and P145L, prevent ligand binding. A two-amino-acid insertion at residues 161 and 162, as well as a T176I missense mutation, also affect ligand binding.96,97,98 A L183P mutation, which is near to, but not in the loop containing Y190, affects both receptor expression and function.99
Mutations in Integrin αIIbβ3 That Affect Receptor Activation Several β3 subunit missense mutations (C560R, V193M) result in the receptor adopting a high-affinity ligand binding state, which is paradoxical as it results in a bleeding diathesis.100,101 A β3 subunit S527F mutation in the third I-EGF domain was also associated with a constitutively active receptor, presumably because it prevents the receptor from assuming a bent, inactive conformation.102 The cytoplasmic domain of the β3 subunit plays a functional role in integrin activation and the regulation of ligand binding.103,104 Two GT mutations have been identified in this region. One is an R724X nonsense mutation (patient RM)105 that results in the deletion of the carboxyterminal 39 residues of integrin β3 and the other is a β3 subunit S752P missense mutation (patient P or Paris I).106,107,108 This latter patient is unusual in that he had a generally mild history of excessive hemorrhage, but he did have a prolonged bleeding time and his platelets did not aggregate in response to ADP. These mutations do not severely affect surface expression of platelet integrin αIIbβ3 complexes, but both mutant receptors are unresponsive to agonist stimulation. Mammalian cell expression studies of these mutations show normal adhesion to immobilized fibrinogen, but abnormal cell spreading. Cells expressing the S752P mutant receptors have reduced focal adhesion plaque formation and cells expressing the R724X mutant receptors have undetectable tyrosine phosphorylation of focal adhesion kinase, pp125FAK. These mutations provide evidence for the role of the β3 subunit cytoplasmic tail in inside-out signaling (i.e., platelet signals that lead to integrin αIIbβ3 adopting a high-affinity ligand-binding conformation) and outside-in signaling (i.e., signaling to the interior of the platelet as a result of integrin αIIbβ3 binding ligand; see Chap. 112, Figs. 112–3, 112–4, and 112–12).
Variants of Integrin αIIbβ3 in the Population The application of missense variant whole-exome and whole-genome sequencing to large numbers of individuals has provided valuable information on the frequency of missense variants in the general population and the frequency of the genetic alterations leading to GT. For the most part, the frequency of a variant in a population is a reflection of its impact on reproductive fitness and when it entered the population, with lower frequencies for variants that entered the population more recently. Thus, variants with minor allele frequencies (MAFs) of approximately 0.5 percent or less probably entered the population fewer than 2500 years ago, when the recent explosive growth in human populations began.109 Data from a study109A involving approximately 33,000 alleles from approximately 16,500 people demonstrated the presence of 114 novel missense variant affecting approximately 10 percent of the integrin αIIb amino acids and approximately 9 percent of the β3 subunit amino acids. Thus, approximately 1.1 percent of the population studied carried at least one missense variant. None of the known GT mutations was observed in any of the alleles studied, indicating that they have MAFs of less than 0.01 percent and thus entered the population very recently. In fact, studies of two GT populations with high intragroup marriages, Palestinian Arabs and French Manouche gypsies, estimated that the GT mutations entered the population approximately 300 to 600 and approximately 300 to 400 years ago, respectively.110,111 Several novel missense variants identified in this study affected one of the amino acids mutated in patients with GT, and in two cases these variants were shown to profoundly affect expression of the receptor. In one case, a missense variant reduced expression by approximately 50 percent but did not alter function. A series of prediction tools indicated that somewhere between 45 and 74 percent of the 114 novel missense mutations may be deleterious. Thus, perhaps approximately 0.6 percent of individuals in the general population is a silent carrier for a GT variant that profoundly affects structure and/or function. In addition, some of the rare individuals in the healthy population with levels of integrin αIIbβ3 receptor expression intermediate between those of obligate GT carriers and normal individuals may reflect heterozygosity for “hypomorphic” variants that partially affect receptor expression, but not function.112
Table 120–2 summarizes the clinical manifestations of 177 patients with GT obtained from two reviews.22,33 Menorrhagia occurs in nearly all female patients. Purpura can be present immediately after birth but often is not dramatic. Petechiae of the face and subconjunctival hemorrhage associated with crying may be the first symptoms in neonates and babies. Spontaneous hemarthroses and central nervous system bleeding are very rare. The hemorrhagic diathesis in patients with GT is notable for its variability and the lack of correlation between the biochemical platelet abnormalities and clinical severity.22 Even within groups of patients such as the Iraqi Jews, most of whom share the same genetic αIIb or β3 subunit abnormalities, there is a wide spectrum of clinical severity.33,39 Moreover, the severity of bleeding symptoms can vary significantly during the lifetime of individual patients. GT does not appear to protect against the development of atherosclerosis as judged by the carotid artery intima-to-media ratio.113 Carriers of GT are usually asymptomatic or only mildly symptomatic and generally have normal results in platelet function tests.22,33,112,114,115
| No. of Affected Patients | Frequency (%) | |
|---|---|---|
| SYMPTOMS | ||
| Menorrhagia | 54/55 | 98 |
| Easy bruising, purpura | 152/177 | 86 |
| Epistaxis | 129/177 | 73 |
| Gingival bleeding | 97/177 | 55 |
| Gastrointestinal hemorrhage | 22/177 | 12 |
| Hematuria | 10/177 | 6 |
| Hemarthrosis | 5/177 | 3 |
| Intracranial hemorrhage | 3/177 | 2 |
| Visceral hematoma | 1/177 | 1 |
| SEVERITY | ||
| Requirement for red cell transfusions | ||
| Patients from literature* | 32/48 | 67 |
| Paris patients | 54/64 | 84 |
Table 120–3 provides characteristic laboratory findings in GT. Patients have normal platelet counts and morphology, prolonged bleeding times, decreased or absent clot retraction, and abnormal platelet aggregation responses to physiologic stimuli. The initial slope of high-dose ristocetin-induced aggregation is normal (or near normal), reflecting the normal plasma VWF and the normal platelet GPIb/IX content; at lower doses of ristocetin, however, where GPIb/IX–mediated activation of integrin αIIbβ3 (Chap. 112) normally contributes to the aggregation response, patients have decreased second wave aggregation.116 GT platelets undergo normal shape change in response to ADP and thrombin, demonstrating their ability to undergo metabolic and cytoskeletal changes in response to these agents. Similarly, high doses of thrombin and collagen produce normal release of dense body and α-granule contents18,20,117; the decreased secretion observed with lower doses of these agents reflect the lack of augmentation of the release reaction normally produced by platelet aggregation.18,116,118,119,120
|
Platelets in whole blood or platelet-rich plasma adhere to glass because fibrinogen first becomes deposited on the glass and the platelets then adhere to the immobilized fibrinogen.121,122 Platelets from patients with GT fail to adhere to glass,18,20,121 and this forms the basis of their abnormality in the glass bead retention assay.123 Platelet coagulant activity has been variably reported as normal or abnormal.18,19,20,21,124,125,126 A defect in platelet microparticle formation and support of thrombin generation has been identified in some patients,125,126,127,128 but not in all patients.129 Integrins αIIbβ3 and αVβ3 bind prothrombin, probably accounting for some of the abnormalities identified.130,131
In flow-chamber studies, thrombasthenic platelets adhere normally to deendothelialized blood vessels at low and intermediate shear rates, but do not spread normally or form platelet thrombi.132,133,134 A defect in adhesion occurs at higher shear rates. A paradoxical increase in fibrin formation on these surfaces has been observed with thrombasthenic platelets, but the explanation for this phenomenon remains unknown.135 In contrast to normal blood, blood from nearly all patients with GT fails to occlude a 150-μm PFA-100 aperture in collagen-coated membranes under high sheer, either in the presence of ADP or epinephrine.136,137
Platelet integrins αIIbβ3 and αVβ3 can be quantitated by several techniques, including, monoclonal antibody binding (using flow cytometry or radiolabeled binding), immunoblotting, and surface-labeling followed by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE). Based on such studies, GT patients are subcategorized by integrin αIIbβ3 content into those with less than 5 percent of normal (type I), 5 to 20 percent (type II), or 50 percent or more (variants).22,138 In one review of 64 patients, 78 percent were type I, 14 percent were type II, and 8 percent were variants.22 This subtyping predated the identification of integrin αIIbβ3 abnormalities as the cause of GT and was based on functional data; this categorization provides only limited information.
Measuring integrin αVβ3 content is technically more demanding than measuring that of integrin αIIbβ3 because there are only approximately 50 to 100 integrin αVβ3 receptors per platelet.63 The integrin αVβ3 level is very useful, however, in making a preliminary assessment of whether the patient has a defect in the αIIb or β3 subunits, because, in general, patients who lack integrin αVβ3 receptors have a defect in the β3 rather than αIIb subunit.139 A β3 subunit missense mutation (H280P) that differentially affected integrin αIIbβ3 more than αVβ3 has, however, been described.69
Fibrinogen-binding studies assess the function of the integrin αIIbβ3 complex.25 Early studies used radiolabeled fibrinogen to the binding of fibrinogen when the platelets are stimulated with ADP25 or a similar agonist. Fibrinogen can also be labeled with a fluorescent molecule and then flow cytometry can be used to measure fibrinogen binding. These techniques are most useful in detecting qualitative abnormalities of integrin αIIbβ3 in patients with variant GT. The binding of a monoclonal antibody (PAC1) to platelets gives similar information because the antibody only binds to the activated form of the integrin.140
Carriers of GT have essentially normal platelet function.34 Their platelets, however, only contain approximately 60 percent of the normal number of integrin αIIbβ3 receptors; the overlap in values between normal individuals and carriers, however, doesn’t permit for unequivocal diagnosis of carriers by this technique.112 Carrier detection is most accurately performed by DNA analysis.
Platelet fibrinogen is reduced to approximately 10 percent of normal in patients with marked reductions in integrin αIIbβ318,21,43,44 but is variably reduced in patients with significant amounts of integrin αIIbβ3.138,141,142
Therapy of GT patients is discussed in the section entitled “Management of Inherited Platelet Function Disorders.” Although GT is a severe disease, the prognosis for survival is generally good. In one series, two of 64 patients died of hemorrhage and in another series, three of 43 patients died of hemorrhage.22,33 A nationwide survey in Japan identified 98 GT patients in 1976 and 192 in 1991.143 The mortality rate decreased substantially during this time interval.
αIIbβ3: Select Macrothrombocytopenias Five heterozygous missense mutations in four different amino acids in integrin αIIbβ3 (αIIb G991C, R995Q, and R995W, and β3 L718P and D723H), as well as several different deletions, lead to variably mild reductions in both integrin αIIbβ3 expression and platelet aggregation, as well as constitutively active receptors, in patients with inherited aniso- and macrothrombocytopenia.144,145,146,147,148,149,150 The defects cluster on both sides of the transmembrane domains, and include both members of the αIIb R995-β3 D723 salt bridge proposed to maintain the receptor in a low affinity state.151 Proplatelet formation has been reported to be abnormal in several reported cases.
BSS is an inherited disorder of the platelet GPIb–IX–V complex characterized by thrombocytopenia, giant platelets, and a failure of the platelets to bind GPIb ligands, most importantly, VWF and thrombin.152,153,154,155
In 1948 Bernard and Soulier described two children from a consanguineous family who had a severe bleeding disorder characterized by mucocutaneous hemorrhage, variable thrombocytopenia, and giant platelets.156,157 Beginning in the early 1970s, BSS platelets were shown to have a functional defect in VWF-dependent platelet adhesion and agglutination.158,159,160 In 1975, Nurden and Caen identified an abnormality in platelet GPIb as the cause of the functional defect.161 Later studies confirmed the defect in VWF–GPIb interactions162,163,164 and identified additional defects in platelet GPV and GPIX.165,166 Subsequent studies have identified additional ligands for the GPIb–IX complex, including thrombin,167 P-selectin,168 leukocyte integrin αMβ2,169 high-molecular-weight kininogen,170 thrombospondin-1,171 and coagulation factors XI172 and XII173 (Chap. 112), but the precise contributions of these interactions to the disorder are not well defined. Molecular defects in the genes for GPIbα, GPIbβ, and GPIX, but not GPV have been identified in BSS. Mouse models of BSS have been produced by gene targeting of GPIbα174 and GPIbβ,175 and like humans, mice deficient in GPV do not demonstrate the typical features of human BSS.176,177
This rare disease, with a prevalence estimated as less than one in 1,000,000, has been reported from countries around the world.152,154,157,165 Both autosomal recessive (“biallelic”) and autosomal dominant (“monoallelic”) forms of the disorder have been described, with the biallelic producing the most severe symptoms and the monoallelic causing macrothrombocytopenia and a mild or no bleeding syndrome. Consanguinity is common in the biallelic form, with 85 percent of the reported cases being homozygous for the causative mutation.154
Six different features of BSS may contribute to the hemorrhagic diathesis: thrombocytopenia, abnormal platelet adhesive interactions with VWF, abnormal platelet interactions with thrombin, abnormal platelet coagulant activity, abnormal platelet interactions with P-selectin, and abnormal platelet interactions with leukocyte integrin αMβ2.
The pathophysiology of the thrombocytopenia is uncertain. Early studies suggested a marked shortening of platelet survival, presumably from the decrease in platelet surface charge resulting from the GPIb defect.178,179 Later studies using 111In-oxine to label platelets reported more modest or no shortening of platelet survival, indicating that ineffective thrombopoiesis and/or decreased thrombopoiesis may contribute to the thrombocytopenia.180,181 Morphologic abnormalities have been identified in BSS megakaryocytes and these may contribute to abnormal platelet production.182 Based on observations in other giant platelet syndromes (Chap. 117), the large size of Bernard-Soulier platelets would tend to diminish the adverse hemostatic effects of the thrombocytopenia because the platelet mass is better preserved. With only rare exceptions,183 however, the bleeding diathesis with BSS is more severe than expected from the thrombocytopenia, reinforcing the conclusion that a qualitative platelet defect is the predominant problem.157,184
The platelet GPIb–IX complex functions as a receptor for VWF (Chaps. 112 and 126).152,185,186 This interaction is crucial in the adhesion of platelets to subendothelial surfaces, especially under high shear conditions, where VWF acts as a bridge between the subendothelial matrix and the platelet.133,134 The relative roles of subendothelial VWF, plasma VWF, and platelet VWF have not been completely defined, but they probably all contribute to platelet adhesion.187 The interaction of VWF with GPIb/IX initiates activation of integrin αIIbβ3,188,189 which can also bind to VWF, but at a different site on the molecule.190 The interaction of GPIb/IX with VWF also directly contributes to platelet–platelet interactions.191,192,193
GPIb/IX–VWF interactions can also occur in platelet suspensions at high shear rates; this can lead to platelet activation, with subsequent aggregation mediated by integrin αIIbβ3.187,194,195,196 Whether sustained shear rates in vivo ever reach the levels required to initiate VWF binding, however, is not established.
Abnormalities of the GPIb–IX complex can be a result of genetic defects in GPIbα, GPIbβ, or GPIX, all of which are required for surface expression. BSS is the most severe form of the disease and is caused by defects in both alleles of one of the proteins as a result of a homozygous mutation, compound heterozygosity, or a combination of hemizygosity of GPIbβ because of a microdeletion and a mutation affecting the other GPIbβ allele. These abnormalities have been termed the biallelic forms.154 A macrothrombocytopenic syndrome associated with a mild bleeding syndrome has been reported with heterozygous defects in GPIbα and GPIbβ.154 Because obligate heterozygotes for the biallelic BSS mutations do not commonly demonstrate macrothrombocytopenia, the heterozygous defects associated with macrothrombocytopenia may exert a dominant negative effect.154
The platelets of patients with BSS have a decreased response to platelet activation by thrombin, especially at limiting concentrations of thrombin.197,198,199 BSS platelets are deficient in two different proteins that interact with thrombin, namely GPIbα, which binds thrombin,167 and GPV, which is a thrombin substrate (Chap. 112). The precise nature of the interactions of thrombin with GPIbα and its biologic consequences are still unclear, but binding of thrombin to GPIbα can initiate signaling within the platelet, perhaps directly through GPIbα crosslinking or indirectly by augmenting activation of other thrombin receptors (protease-activated receptors [PARs] 1 and 4) or other thrombin-dependent events at the platelet surface.167 Paradoxically, mice deficient in GPV actually have increased sensitivity to thrombin activation and variably increased thrombus formation, perhaps because GPV limits access of thrombin to GPIb.200,201 Because thrombin is one of the major physiologic activators of platelets, the loss of thrombin binding to GPIbα may contribute to the hemorrhagic diathesis.
Platelets from patients with BSS are defective in supporting thrombin generation as judged by the serum PT,202 a test performed with whole blood, but in other tests of platelet coagulant activity, BSS platelets support coagulation as well as, or better than, normal platelets.124,203 Defects in collagen-induced coagulant activity and the association of factors V, VIII, and XI with BSS platelets have been described,203 but their significance is unclear. Similarly, GPIb/IX has been identified as a binding site for other proteins involved in coagulation, including high-molecular-weight kininogen and factor XII, but the contributions of these interactions to the coagulant abnormality are also uncertain.170,172,173 Binding of VWF to GPIb/IX has been implicated in fibrin-dependent, but not fibrin-independent, augmentation of platelet coagulant activity and thus fibrin-dependent coagulant activity is likely to be abnormal in BSS.126 This finding may partially explain the variability in findings between the serum PT and some of the other assays as fibrin only forms in the serum PT. Abnormal membrane lipids have also been reported.204
The mechanism(s) producing the giant platelets in BSS has not been identified, but since giant platelets are found in BSS variants in which GPIb/IX is present, but unable to bind ligand, it has been postulated that the abnormality is a result of the inability of GPIb/IX to bind an unknown marrow ligand.152 It cannot be because of an inability to bind VWF as, with only rare exceptions,205 patients lacking VWF do not have large platelets. Moreover, in a mouse model of BSS, restoring a receptor with the GPIb transmembrane and cytoplasmic domains, but not the ligand-binding domain, partially corrected both the thrombocytopenia and large platelet size.206 A defect in GPIb/IX–mediated signaling has also been proposed to cause the large platelets as a deficiency of PLC has also been described in BSS.152,207 A mechanical alteration in the plasma membrane of BSS platelets has been identified by micropipette experiments, showing the plasma membrane to be more deformable than normal.208 Megakaryocytes in BSS have increased ploidy and volume, as well as alterations in the membrane demarcation system, granules, and microtubules.181,182 Both the increased size and deformability may reflect the loss of the normal interaction of GPIb/IX with the cytoskeleton via actin-binding protein (filamin-1; Chap. 112).
Platelets from patients with BSS are deficient not only in GPIbα, GPIbβ, and GPIX, which are known to be associated as a complex, but also in GPV (Chap. 112).152,166 All of these proteins share highly conserved leucine-rich regions.152,187 One possible explanation for the loss of surface expression of all the proteins is that they need to form a complex during biosynthesis in order to be transported to the surface187; evidence supports the need for GPIbα, GPIbβ, and GPIX to all be present for optimal surface expression,209 but data from mice deficient in GPV indicate that this glycoprotein is not required for surface expression of the GPIb–IX complex.200 GPV may, however, improve the efficiency of expression of the other members of the complex.210 Moreover, data from the BSS mouse expressing a chimeric GPIbα molecule in which the leucine-rich repeat domain was replaced with the external domain of another receptor indicate that complex formation does not require the GPIbα leucine-rich domain.206
At the molecular level, the platelets from different patients with BSS are heterogeneous, with many having no detectable GPIb and others having variable amounts, up to 50 percent of normal.152,207,211,212,213,214 There also is variability in the degree of concordance in the reduction of GPIb and the other deficient proteins.215,216
Molecular Defects The molecular biologic basis of BSS has been determined in 161 patients from 132 unrelated families154 and an online registry of defects is available at http://www.bernardsoulier.org/.217 An international consortium reported on 211 families with the recessive form of BSS, which they termed “biallelic.”154 In total, 45 different mutations have been reported in GPIbα, 52 in GPIbβ, and 28 in GPIX. No defects in GPV have been identified in patients with BSS. The association with consanguineous matings was reinforced as 85 percent of the families had homozygous mutations and 13 percent were compound heterozygotes for defects in one of the genes. None of the variants were identified in several gene variant databases,154 suggesting that they are all rare and likely entered the population relatively recently. A number of likely founder mutations have been identified in each of the three genes in different populations.154,218 The ancestry of seven apparently unrelated families with a GPIbβ W89D mutation was traced to a common ancestor in 1671 in India.218 Five mutations in GPIX account for 137 of the 184 affected GPIX alleles and GPIX N61S is found in 64 European families. A number of mutations have been identified to cause the heterozygous monoallelic form, including the GPIbα A172V mutation, which has been associated with biallelic and monoallelic forms of the disorder in 42 apparently unrelated families with macrothrombocytopenia in Italy.154 Many of the defects affect the leucine-rich repeats or the conserved flanking sequences, supporting the importance of these structural elements in the biogenesis and surface expression of the GPIb–IX–V complex (see Chap. 112, Fig. 112–14, and Fig. 120–4). Three patients have been described who are homozygous for a deletion in the last two bases of codon 492 of GPIbα, resulting in a frameshift that alters the membrane spanning region and results in premature termination, and another patient has been described who is heterozygous for this deletion and a missense mutation of GPIbα.219,220,221,222 These defects appear to result in a poorly anchored GPIbα with GPIbα antigen present in plasma. GPIbβ mutations have affected the promoter region, at a binding site for the GATA-1 transcription factor,223 the signal peptide,224 and the transmembrane and intracellular domains.225 A homozygous Y88C defect in GPIbβ has been reported to cause BSS in two Japanese families and heterozygotes with this mutation have a giant platelet syndrome.221,226 Similarly, a patient heterozygous for a GPIbβ R17C mutation also had a giant platelet syndrome.227 An N45S mutation in GPIX, affecting leucine rich repeat 1, has been reported in at least 12 different white patients, including four patients from a large Swiss family with variable clinical manifestations228,229,230 and one Turkish patient.231
Figure 120–4.
Localization of select missense mutations causing platelet-type von Willebrand disease (VWD) and Bernard-Soulier syndrome (BSS) in the GPIbα N-terminal domain. Ribbon diagram of the topology of GPIbα N-terminal domain viewed from the side. The regulatory loop is colored blue with activating platelet-type VWD mutations G233V and M239V indicated as open black balls. Five BSS mutations, which cause loss of von Willebrand factor binding, are shown as blue balls. L57F and C65R localize to leucine-rich repeat (LRR) 2 with L129P, A156V, and L179del localized to the LRR5, LRR6, and LRR7 β-strands, respectively. The molecular structure of the sulfated tyrosine residues 276, 278, and 279 are shown. (Adapted with permission from Uff S, Clemetson JM, Harrison T, et al: Crystal structure of the platelet glycoprotein Ib(alpha) N-terminal domain reveals an unmasking mechanism for receptor activation. J BiolChem 277(38):35657–35663, 2002.)
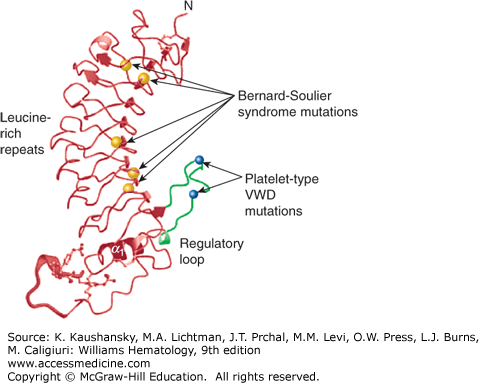
BSS has been reported in seven patients in association with hemizygous deletion of GPIbβ and several neighboring genes on chromosome 22q11.2, leading to variable manifestations of the DiGeorge syndrome, including cardiac defects, dysmorphic facial features, thymic hypoplasia, and velopharyngeal insufficiency.154,232–239 Hemizygous mutations in the remaining GPIbβ allele have included P96S and P29L.236,237 In other studies of patients with the 22q11.2 deletion syndrome, modest reductions in platelet count and increases in platelet volume, as well as reduced platelet agglutination to ristocetin and decreased platelet GPIb/IX expression, have been variably reported, consistent with hemizygosity for GPIbβ.240,241,242,243,244
A number of monoallelic, heterozygous mutations in the genes of the GPIb–IX complex have been described as causing macrothrombocytopenia, some, but not all, of which have also been implicated in causing the biallelic form either because of homozygosity or compound heterozygosity.154 These include a heterozygous mutation in the second leucine-rich repeat (L57F)245 in which the affected patients have moderate bleeding symptoms, moderate thrombocytopenia, and giant platelets. Additional monoallelic mutations of GPIbα that appear to produce dominant effects are N41H246 and Y54D.247
The “Bolzano” defect, which involves a mutation in the sixth leucine-rich repeat of GPIbα (A156V), results in a GPIbα molecule that has reduced ability to bind VWF, but can bind thrombin. It has been described in both biallelic and monoallelic forms. Two patients with biallelic forms have been described. In one patient, the Bolzano defect was homozygous, and the patient had a lifelong history of mucocutaneous hemorrhage in association with an approximately 50 percent reduction in GPIb surface expression and total loss of ability to bind VWF.248 In the other, the Bolzano mutation coexisted with a 12-amino-acid deletion and an amino acid substitution (Q181K).249 The monoallelic form of the Bolzano defect has been reported in more than 100 patients from 48 pedigrees, primarily from southern Italy.250 Most patients have no bleeding symptoms, but some have mild to moderate bleeding symptoms. Mild thrombocytopenia, increased mean platelet volume, reduced expression of GPIb/IX/V, and normal or borderline abnormal values for ristocetin-induced platelet aggregation are characteristic of this disorder.
Epistaxis is the most common symptom of BSS (70 percent); also common are ecchymoses (58 percent), menometrorrhagia (44 percent), gingival hemorrhage (42 percent), and gastrointestinal bleeding (22 percent).157 The combination of BSS with angiodysplasia can result in particularly severe recurrent hemorrhage.251,252,253 Hemorrhagic symptoms that occur with lower frequency include posttraumatic bleeding (13 percent), hematuria (7 percent), cerebral hemorrhage (4 percent), and retinal hemorrhage (2 percent). There is considerable variability in symptoms among patients, even among patients within a single family.152,254 A review that includes brief descriptions of the clinical features of 55 patients, reported through 1998, has been published.152
Thrombocytopenia is present in nearly all patients, but is variable in its severity, ranging from approximately 20 × 109 platelets/L to near-normal levels. Platelets are large on smear, with more than one-third usually having diameters greater than 3.5 μm, and some being as large or larger than lymphocytes. By electron microscopy, platelets display only minor variations in vesicular structures and the open canalicular system,157 but megakaryocytes have more notable abnormalities in their demarcation membranes.182 The cell membranes of platelets from patients with BSS appear to be more deformable than normal,208 perhaps because GPIb ordinarily interacts with the platelet cytoskeleton255 (Chap. 112).
Closure times of the apertures of collagen-coated membranes are markedly prolonged in the presence of ADP or epinephrine (PFA-100).136 The hallmark findings in the BSS are the failure of platelets to aggregate in response to ristocetin159 or botrocetin,162,256 agents that require VWF–GPIb interactions. In VWD, but not BSS, this defect can be corrected by adding normal plasma (or VWF).
Although, the large size of the platelets in BSS and the thrombocytopenia make it technically difficult to perform platelet aggregation studies, in general, aggregation induced by ADP, epinephrine, or collagen is either normal or enhanced.160,257,258 The aggregation response to thrombin is usually dose-dependent, being essentially normal in response to high doses of thrombin but characterized by a prolonged lag phase and diminished aggregation in response to low doses of thrombin.197,259
Platelet Coagulant Activity The coagulant activity of platelets from patients with BSS has been variably reported as reduced, normal, or increased.124,202,203 The variable presence of fibrin in the different assays used to assess platelet coagulant activity may account for these inconsistent results as GPIb–VWF interactions enhance platelet coagulant activity when fibrin is present, but not when it is absent.126
Platelet-Thrombin Interactions Both GPIb and the seven-transmembrane domains PAR-1 and PAR-4 receptors are required for maximal response to thrombin.167,259 Two different crystal structures of the interactions between thrombin and GPIbα have been reported; in one, two molecules of thrombin bind to each GPIbα molecule, raising the possibility that free thrombin or thrombin adherent to fibrinogen can cluster GPIb–IX–V complexes.167,260,261 GPV, which is missing from the platelet surface in BSS, is cleaved by thrombin, but the cleavage is neither necessary nor sufficient for thrombin-induced platelet activation.262,263 In fact, platelets of mice lacking GPV have increased responsiveness to thrombin, perhaps because GPV ordinarily limits access of thrombin to GPIbα or inhibits GPIbα crosslinking.200,201
Ex Vivo Interaction with Subendothelial Surfaces Platelets frompatients with BSS demonstrate defective adhesion to subendothelial surfaces, especially at shear rates greater than 650 s–1.133,134,158,264 The results are similar to those in patients with VWD.
Shear-Induced Platelet Aggregation Unlike normal platelets, platelets from patients with BSS are not aggregated by high shear rates.194,195 The initial interaction in this process appears to be binding of VWF to GPIb,187 with subsequent activation of integrin αIIbβ3. perhaps through signaling via the protein 14-3-3ζ associated with the cytoplasmic domain of GPIbα,196,265 Fcγ receptor IIA, GPVI, and/or the Fc receptor γ chain (Chap. 112).266,267,268 Pathologic shear stress has been reported to increase binding of α-actin to GPIb/IX as part of the signaling process.268,269,270
The therapy of BSS is described in the section “Management of Inherited Platelet Function Disorders” below. Splenectomy has been performed when the diagnosis of immune thrombocytopenia was mistakenly made, but this usually does not normalize the platelet count or improve the bleeding diathesis.249
A heterogeneous group of patients has been described with mild to moderate bleeding symptoms, variably enlarged platelets, variable thrombocytopenia, and diminished plasma high-molecular-weight VWF multimers.271 The fundamental defect in these patients is thought to be an enhanced interaction between an abnormal platelet GPIb/IX receptor and normal plasma VWF.272–281 Because these patients have some of the hallmarks of VWD, but the defect is in platelet GPIb/IX, it has been termed both pseudo-VWD and platelet-type VWD. At present, 55 patients are listed in the database of patients with this disorder (www.pt-vwd.org).282,283
A qualitative abnormality in GPIb is responsible for this disorder, with ongoing in vivo binding of high-molecular-weight VWF multimers to platelets causing depletion of the plasma high-molecular-weight multimers. In addition, the binding of the VWF to platelets may lead to shortened platelet survival, perhaps accounting for the variable thrombocytopenia. Inheritance is autosomal dominant.
Abnormalities in the Mr of GPIb were identified in two families,277 but these may have resulted from a now-recognized polymorphism in GPIb (Chap. 112) rather than being related to the functional disorder. Heterozygous point mutations in the GPIbα gene causing a variety of missense alterations (G233V, G233S, M239V, D235Y, W230L) have been found in several different families.271,278,284,285,286,287,288,289 The G238V mutation is the most common among the patients in the database, affecting 31 of 35 patients. All of these mutations are in the R-loop (also termed β-switch) in the carboxyterminal flanking sequence of the leucine-rich repeats, a region implicated in ligand binding (see Fig. 120–4).152,187,290,291,292,293 Molecular modeling suggests that the M239V substitution produces a significant conformational change in the molecule,294 and this was confirmed by crystallographic analysis.295 It has been proposed that the platelet-type VWD mutations either destabilize the compact triangular structure of the R-loop by interfering with the D235-K237 salt bridge or stabilizing the extended β-hairpin form of the R-loop, which is better able to engage the VWF A1 domain.282 A mouse model of the GPIbα G233V mutation recapitulated many of the human manifestations and had an unexpected increase in bone mass.296 Recombinant GPIbα fragments containing the G233V and M239V mutations demonstrated enhanced interactions with VWF in several different systems, including ones under shear stress.297,298 An increase in platelet GPIb/IX expression has also been reported.278,281 An in-frame 27-base-pair (bp) deletion in the macroglycopeptide region of GPIbα has also been reported to cause platelet-type VWD.279 Because the deletion may lead to the loss of up to four glycosylation sites, it has been proposed that the glycans play a negative regulatory role in ligand binding.282
Patients have variable thrombocytopenia and mild to moderate mucocutaneous hemorrhage. A study of 13 patients with six different mutations using a standardized bleeding assessment tool found a wide range of clinical severity, with approximately 40 percent having a normal bleeding score and the remainder having a wide range of abnormal scores.271 Bleeding scores did not correlate with age or sex, but did correlate with reductions in both platelet count and ristocetin cofactor activity. All patients had macrothrombocytopenia. Of note, pregnancy may exacerbate the thrombocytopenia and bleeding symptoms.278
Mild thrombocytopenia and somewhat enlarged platelets are present in some, but not all, patients. Plasma VWF levels are variably reduced, with a disproportionate reduction in plasma high-molecular-weight multimers. Platelet VWF multimers are normal.
The most characteristic laboratory finding in platelet-type VWD is enhanced platelet aggregation in response to low concentrations of ristocetin272,273,274,275,276,278,288 or botrocetin.299 This same abnormality is present in patients with type 2b VWD, as is selective depletion of plasma high-molecular-weight VWF multimers (Chap. 126). In platelet-type VWD, however, the defect is in platelet GPIbα, whereas in type 2b VWD, the defect is in the VWF. In one study comparing platelet-type and type 2b VWD, patients with type 2b VWD had more severe bleeding, especially menorrhagia, and lower platelet counts.271 Several assays can help differentiate between these abnormalities274,300,301,302: (1) normal VWF (purified or in cryoprecipitate) will aggregate platelets from patients with platelet-type VWD, but not platelets from patients with type 2b VWD; (2) isolated platelets from patients with platelet-type VWD will bind normal VWF at lower concentrations of ristocetin than will normal platelets or platelets from patients with type 2b VWD; (3) plasma VWF from patients with type 2b VWD will bind to normal platelets at lower-than-normal concentrations of ristocetin, whereas higher-than-normal concentrations of ristocetin are required to promote the plasma VWF from patients with platelet-type VWF to bind to normal platelets301; and (4) VWF lacking sialic acid residues (asialo-VWF) will agglutinate platelets from patients with platelet-type VWD in the presence of ethylenediaminetetraacetic acid (EDTA).303 A number of patients with platelet-type VWD were originally diagnosed as having type 2b VWD, leading to the conclusion that platelet-type VWD may be underdiagnosed, and an international registry based study supports this contention.278,280,304
Because normal VWF (especially the high-molecular-weight forms) can bind excessively to the platelets of patients with platelet-type VWD and potentially lead to rapid platelet clearance from the circulation, increasing the VWF level by any means (desmopressin infusion or VWF replacement with cryoprecipitate or VWF concentrates) poses a potential risk of inducing thrombocytopenia.300,305 It may be possible to estimate this risk by assessing whether the patient’s platelets aggregate ex vivo in response to VWF (as in cryoprecipitate).273 Low-dose cryoprecipitate has successfully supported hemostasis, without inducing thrombocytopenia.275,305,306 Currently, cryoprecipitate is generally less favored for VWF replacement therapy than plasma-derived factor VIII concentrates such as Humate-P, which is approved in the United States for the therapy of VWD, because the plasma-derived factor VIII concentrates have a reduced risk of viral infection. Consideration should also be given to platelet transfusion in appropriate circumstances. Recombinant factor VIIa infusion may be beneficial and is licensed for this indication in Europe, but this therapy is not yet approved by the FDA; it has the theoretical advantage of avoiding excessive interactions between VWF and the abnormal GPIbα receptor.307,308
Integrin α2β1 (GPIa/IIa) can mediate platelet adhesion to collagen and platelet activation under certain conditions (Chap. 112). A female patient with excessive posttraumatic bruising and menorrhagia but no epistaxis, gum bleeding, or excessive bleeding after tonsillectomy or appendectomy was described whose platelets selectively failed to aggregate or undergo shape change in response to collagen.309,310 The bleeding time was markedly prolonged, and the patient’s platelets failed to adhere and spread normally on subendothelial surfaces. The patient’s platelets only contained approximately 15 to 25 percent of the normal amount of integrin α2,309,311 and a reduction in the β1 subunit was also apparent.309 It is difficult to draw conclusions about the physiologic role of integrin α2β1in platelet function from this patient because her α2β1 deficiency was incomplete, her bleeding symptoms were mild and variable, and some of the platelet function abnormalities (e.g., abnormal platelet-collagen interactions in the presence of the divalent chelating agent EDTA) are difficult to ascribe to the deficiency in α2β1.309,312
Another patient with integrin α2 deficiency has been described.313 She had a history of mucocutaneous and postoperative bleeding. Her bleeding time was prolonged and platelet aggregation in response to collagen was selectively reduced, but not absent. In addition to her α2 subunit defect, she also had little or no intact thrombospondin, and exogenous thrombospondin corrected the defect in platelet aggregation. The patient’s hemorrhagic symptoms and platelet defects disappeared when she entered menopause.
The variation in platelet integrin α2β1 expression in healthy individuals is very wide (10-fold) and platelet levels have been correlated with allelic variants.314 Reduced α2
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


