Despite decades of scientific and clinical research, pancreatic ductal adenocarcinoma (PDAC) remains a lethal malignancy. The clinical and pathologic features of PDAC, specifically the known environmental and genetic risk factors, are reviewed here with special emphasis on the hereditary pancreatic cancer (HPC) syndromes. For these latter conditions, strategies are described for their identification, for primary and secondary prevention in unaffected carriers, and for disease management in affected carriers. Nascent steps have been made toward personalized medicine based on the rational use of screening, tumor subtyping, and targeted therapies; these have been guided by growing knowledge of HPC syndromes in PDAC.
Key points
- •
Up to 10% of patients with pancreatic ductal adenocarcinoma (PDAC) have an affected first-degree relative, implying inherited predisposition.
- •
In approximately 10% of these patients, a pathogenic germline variant in a hereditary pancreatic cancer (HPC) syndrome gene can be identified.
- •
Taking a detailed family history is key identifying patients who may have pathogenic germline variants.
- •
A significant number of patients with germline predisposition will not have significant family history because of variable penetrance of some pathogenic variants. Strategies of universal testing for germline variants have not been explored in PDAC.
- •
Primary and secondary prevention programs are recommended for carriers of HPC syndromes, but there is little consensus on this subject. Screening for PDAC should be considered investigational.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) remains among the most lethal malignancies despite more than 5 decades of research into its biology and epidemiology. The high mortality from PDAC is attributable to several factors, including its inherently bad biology, that symptoms and signs are nonspecific and occur late in its natural history, the inability to screen for PDAC, and a dearth of knowledge regarding pathogenesis and evolution of PDAC.
Poor outcomes will be improved when standard of care for PDAC is similar to that of other malignancies, such as colon and breast cancer, by, foremost, screening that allows the detection of noninvasive precursors and nonmetastatic neoplasms, second, by tumor subtyping with the rational use of immunohistochemistry and molecular testing; and finally, by more rational chemotherapy.
There is ample evidence that population-based primary and secondary prevention programs have ameliorated outcomes for more common malignancies, including colorectal and breast, and for malignancies with traditionally high mortality, including primary lung adenocarcinoma. In the case of primary lung adenocarcinoma, this has been seen when screening is focused on those with elevated risk for the disease. In the case of colorectal cancer, this has been seen when screening is tailored to those with hereditary predisposition.
Epidemiologic risk factors for PDAC neither individually nor in aggregate greatly increase the risk of developing the disease. Germline variants in any of 15 genes do significantly increase relative risk of developing PDAC, and these are collectively referred to as hereditary pancreatic cancer (HPC) syndromes. Although much work has been done to investigate these syndromes, questions remain regarding their overall and individual prevalence, their penetrance, their impact on tumor biology, and the benefit of cancer screening in carriers of pathogenic germline variants.
Tumor subtyping has been advantageous in the management of other epithelial adenocarcinomas, including breast and colorectal cancer. With the latter, this has been informed by understanding unique high-risk cases with germline predisposition. For example, Lynch syndrome (LS) cases are marked by somatic tumor microsatellite instability, which is both prognostic of outcome and predictive of chemotherapy response in both the germline and the sporadic microsatellite unstable colorectal cancers. There is strong reason to suspect that PDAC patients with germline predisposition also represent subgroups informative for both those with inherited and those with sporadic defects in key driver genes, and that this grouping is predictive of chemotherapy response, as is described later.
Here, PDAC, including natural history and clinical risk factors, and the HPC syndromes, including strategies for their identification and management, are reviewed, and the importance that these syndromes have in improving the understanding and management of PDAC are relayed.
Clinical and epidemiologic overview
The incidence of PDAC is approximately 4,700 per year in Canada and 46,400 in the United States, meaning that the lifetime risk of developing PDAC is roughly 1.5% in North America.
The mortality of PDAC is nearly as high as its incidence, with an estimated 4,400 deaths per year in Canada and 39,600 deaths per year in the United States. Overall survival at 5 years is 7%, the lowest of all cancers and only double of what it was in 1990. From these trends, it is estimated that PDAC will be the second leading cause of cancer death in North America within 10 years.
For these aforementioned statistics to remain so dismal despite decades of research is testament to the inherent malevolence of PDAC biology. Symptoms and signs of PDAC are nonspecific and occur late in its natural history, with the exception of painless jaundice due to tumors in the head of the gland that fortuitously obstruct biliary drainage. Only 15% to 20% of patients present with potentially curable disease, and often these are detected incidentally. The time from invasion to metastasis was initially thought to be on the order of 10 to 15 years, but more recent work suggests that it is much shorter, possibly being acquired as soon as the tumor becomes invasive. PDAC is also highly resistant to systemic therapies, including newer targeted agents.
Staging investigations may include measurements of serum carcinoembryonic antigen (CEA) and CA 19-9, non-invasive imaging including computed tomography (CT), and MRI, and invasive modalities, including endoscopic ultrasound (EUS), endoscopic retrograde cholangiopancreatography (ERCP) with cytologic brushings and endoscopic or percutaneous fine-needle aspiration (FNA), or core biopsies. Non-invasive imaging allows staging according to the American Joint Committee on Cancer TNM (AJCC) system and by the less rigorously defined, 4-tiered system of potentially resectable, borderline resectable, locally advanced, and metastatic, the former 3 largely dictated by the extent of tumor invasion into adjacent vascular structures. A rapid autopsy series from Johns Hopkins implies that 70% of patients develop miliary metastatic disease, 18% develop oligometastatic, and 12% remain locally advanced. In those patients with potentially and borderline resectable disease, en bloc, oncologic resection by either Whipple, distal, or total pancreatectomy is usually attempted first, followed by adjuvant chemotherapy with either gemcitabine or 5-fluorouracil (5-FU)-based chemotherapy, although preoperative neoadjuvant therapy is used in some centers. Patients with borderline resectable and locally advanced tumors are usually offered chemotherapy, sometimes in the context of a clinical trial, with rare downstaging to resectability.
PDAC management should be in specialized centers, because surgical volume and perioperative mortality are strongly correlated. Standard chemotherapy is gemcitabine, although recent trials suggest potentially greater (albeit slight) benefit with FOLFIRINOX or the combination of gemcitabine with either nab-paclitaxel or erlotinib in the metastatic setting. The role of radiation therapy is less clear. When patients are optimally managed, median survival for potentially resectable cases is 18 to 24 months, borderline resectable 14 to 20 months, locally advanced 6 to 12 months, and metastatic 3 to 9 months.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) remains among the most lethal malignancies despite more than 5 decades of research into its biology and epidemiology. The high mortality from PDAC is attributable to several factors, including its inherently bad biology, that symptoms and signs are nonspecific and occur late in its natural history, the inability to screen for PDAC, and a dearth of knowledge regarding pathogenesis and evolution of PDAC.
Poor outcomes will be improved when standard of care for PDAC is similar to that of other malignancies, such as colon and breast cancer, by, foremost, screening that allows the detection of noninvasive precursors and nonmetastatic neoplasms, second, by tumor subtyping with the rational use of immunohistochemistry and molecular testing; and finally, by more rational chemotherapy.
There is ample evidence that population-based primary and secondary prevention programs have ameliorated outcomes for more common malignancies, including colorectal and breast, and for malignancies with traditionally high mortality, including primary lung adenocarcinoma. In the case of primary lung adenocarcinoma, this has been seen when screening is focused on those with elevated risk for the disease. In the case of colorectal cancer, this has been seen when screening is tailored to those with hereditary predisposition.
Epidemiologic risk factors for PDAC neither individually nor in aggregate greatly increase the risk of developing the disease. Germline variants in any of 15 genes do significantly increase relative risk of developing PDAC, and these are collectively referred to as hereditary pancreatic cancer (HPC) syndromes. Although much work has been done to investigate these syndromes, questions remain regarding their overall and individual prevalence, their penetrance, their impact on tumor biology, and the benefit of cancer screening in carriers of pathogenic germline variants.
Tumor subtyping has been advantageous in the management of other epithelial adenocarcinomas, including breast and colorectal cancer. With the latter, this has been informed by understanding unique high-risk cases with germline predisposition. For example, Lynch syndrome (LS) cases are marked by somatic tumor microsatellite instability, which is both prognostic of outcome and predictive of chemotherapy response in both the germline and the sporadic microsatellite unstable colorectal cancers. There is strong reason to suspect that PDAC patients with germline predisposition also represent subgroups informative for both those with inherited and those with sporadic defects in key driver genes, and that this grouping is predictive of chemotherapy response, as is described later.
Here, PDAC, including natural history and clinical risk factors, and the HPC syndromes, including strategies for their identification and management, are reviewed, and the importance that these syndromes have in improving the understanding and management of PDAC are relayed.
Clinical and epidemiologic overview
The incidence of PDAC is approximately 4,700 per year in Canada and 46,400 in the United States, meaning that the lifetime risk of developing PDAC is roughly 1.5% in North America.
The mortality of PDAC is nearly as high as its incidence, with an estimated 4,400 deaths per year in Canada and 39,600 deaths per year in the United States. Overall survival at 5 years is 7%, the lowest of all cancers and only double of what it was in 1990. From these trends, it is estimated that PDAC will be the second leading cause of cancer death in North America within 10 years.
For these aforementioned statistics to remain so dismal despite decades of research is testament to the inherent malevolence of PDAC biology. Symptoms and signs of PDAC are nonspecific and occur late in its natural history, with the exception of painless jaundice due to tumors in the head of the gland that fortuitously obstruct biliary drainage. Only 15% to 20% of patients present with potentially curable disease, and often these are detected incidentally. The time from invasion to metastasis was initially thought to be on the order of 10 to 15 years, but more recent work suggests that it is much shorter, possibly being acquired as soon as the tumor becomes invasive. PDAC is also highly resistant to systemic therapies, including newer targeted agents.
Staging investigations may include measurements of serum carcinoembryonic antigen (CEA) and CA 19-9, non-invasive imaging including computed tomography (CT), and MRI, and invasive modalities, including endoscopic ultrasound (EUS), endoscopic retrograde cholangiopancreatography (ERCP) with cytologic brushings and endoscopic or percutaneous fine-needle aspiration (FNA), or core biopsies. Non-invasive imaging allows staging according to the American Joint Committee on Cancer TNM (AJCC) system and by the less rigorously defined, 4-tiered system of potentially resectable, borderline resectable, locally advanced, and metastatic, the former 3 largely dictated by the extent of tumor invasion into adjacent vascular structures. A rapid autopsy series from Johns Hopkins implies that 70% of patients develop miliary metastatic disease, 18% develop oligometastatic, and 12% remain locally advanced. In those patients with potentially and borderline resectable disease, en bloc, oncologic resection by either Whipple, distal, or total pancreatectomy is usually attempted first, followed by adjuvant chemotherapy with either gemcitabine or 5-fluorouracil (5-FU)-based chemotherapy, although preoperative neoadjuvant therapy is used in some centers. Patients with borderline resectable and locally advanced tumors are usually offered chemotherapy, sometimes in the context of a clinical trial, with rare downstaging to resectability.
PDAC management should be in specialized centers, because surgical volume and perioperative mortality are strongly correlated. Standard chemotherapy is gemcitabine, although recent trials suggest potentially greater (albeit slight) benefit with FOLFIRINOX or the combination of gemcitabine with either nab-paclitaxel or erlotinib in the metastatic setting. The role of radiation therapy is less clear. When patients are optimally managed, median survival for potentially resectable cases is 18 to 24 months, borderline resectable 14 to 20 months, locally advanced 6 to 12 months, and metastatic 3 to 9 months.
Risk factors
Risk factors for PDAC can be divided into 2 groups, namely environmental and genetic, the latter being either low penetrant, common variants, or more highly penetrant, rare variants.
Numerous putative associations of PDAC with lifestyle factors have been reported in older literature. More recently, 2 large groups, the Pancreatic Cancer Case-Control Consortium (PanC4) and the Pancreatic Cancer Cohort Consortium (PanScan), have carried out methodologically rigorous epidemiologic studies on PDAC risk, and their results are concordant. These studies have shown only a few consistent modifiable risk factors for PDAC ( Table 1 ). These studies are briefly reviewed here, relying mostly on the results of the PanC4 analyses in which the authors’ group was involved.
| Factor | Condition | OR (95% CI) |
|---|---|---|
| Smoking | Current smokers | 2.2 (1.7–2.8) |
| Pancreatitis | Diagnosed 2 or more years before PDAC diagnosis | 2.71 (1.96–3.74) |
| Alcohol | >9 drinks/day | 1.6 (1.2–2.2) |
| Obesity | BMI >35 | 1.55 (1.16–2.07) |
| DM2 | Diagnosed 2 or more years before PDAC diagnosis | 1.90 (1.72–2.09) |
| Allergies | Hay fever or animals | 0.73 (0.64–0.84) |
Smoking is the most established environmental risk factor, and smoking cessation is the only evidence-based PDAC preventative measure. Compared with never smokers, PanC4 found an odds ratio (OR) of 1.2 (95% confidence interval [CI] 1.0 to 1.3) for former smokers and 2.2 (95% CI 1.7–2.8) for current cigarette smokers, with increasing risk with increasing number of cigarettes smoked and increasing duration of cigarette smoking up to 40 years. Risk decreased with increasing time since cigarette smoking cessation, returning to baseline 20 years after quitting. There is also evidence that second-hand smoke exposure (OR 1.21 [95% CI 0.60–2.44]) and cigar smoking also increases the risk of PDAC, although inconsistent associations have been found for pipe smoking and smokeless tobacco.
Diabetes mellitus type 2 (DM2) is a risk factor for PDAC. The association is strongest when DM2 is diagnosed within 2 years of PDAC but persists in those diagnosed 2 or more years before PDAC development. The risk is independent of other factors, including body mass index (BMI) and tobacco smoking. Interestingly, risk decreases with duration of diabetes but never reaches baseline, with an OR of 1.30 (95% CI 1.03–1.63) 20 or more years after diabetes diagnosis. Use of oral hypoglycemics for more than 15 years is protective (OR 0.31, 95% CI 0.14–0.69), while insulin use is associated with increased risk (OR 2.66, 95% CI 2.07–3.43), although not for 10 or more years’ duration, implying reverse causation and strongly linked covariates in this association. Thus, although DM2 diagnosed within 2 years of a PDAC diagnosis may be a consequence of the growing neoplasm (so-called Type3c DM), there is little doubt that diabetics are at increased risk for PDAC, the biological mechanism of which may be increased circulating levels of mitogenic insulin due to peripheral insulin resistance.
Pancreatitis and PDAC are associated conditions. The strength of the association is less at diagnostic intervals of greater than 2 years (OR: 2.71, 95% CI: 1.96–3.74) compared with intervals of less than 2 years (OR: 13.56, 95% CI: 8.72–21.90), probably due to both reverse causation and misdiagnosis of PDAC as pancreatitis. At intervals of greater than 2 years, the population attributable fraction is only 1.34% (95% CI: 0.612%–2.07%). Thus, while the inflammation of pancreatitis does likely predispose to PDAC, it accounts for only a small proportion of cases.
There is no association between occasional (<1 drink per day) and moderate (≤4 drinks per day) alcohol consumption and PDAC risk. Heavy alcohol consumption (≥6 drinks per day) does increase risk, independently of type of drink, duration of drinking, tobacco smoking, history of pancreatitis, or race.
Severe obesity (BMI >35) and risk of PDAC are positively associated according to a PanScan study (OR 1.55 [95% CI 1.16–2.07]). Although this effect may have been attenuated when controlling for DM2 status, there was nonetheless a significant trend for increasing PDAC risk with increasing BMI (adjusted OR for the highest vs lowest BMI quartile, 1.33; 95% CI, 1.12–1.58; P (trend) <.001). A separate meta-analysis has shown that the estimated summary relative risk of PDAC per 5 point increase in BMI was 1.12 (95% CI 1.06–1.17), and that this was essentially independent of gender.
Previous associations of PDAC with peptic ulcer disease and recent surgery, especially gastrectomy and cholecystectomy, have been disproved by PanC4 analyses. The associations were strongest within 2 years of PDAC diagnosis with no significantly increased risk observed beyond 2 years, strongly suggesting that the previously observed associations were due to increased cancer detection during investigation, misdiagnosis of symptoms caused by PDAC, and treatment of those other conditions.
The authors’ group and others have recently provided evidence for a protective association between allergies and PDAC risk. Respiratory allergies, especially hay fever (OR = 0.74, 95% CI: 0.56, 0.96), and allergies to animals (OR = 0.62, 95% CI: 0.41, 0.94) are most related to lower risk, while other allergies and asthma may not be protective. Older age at onset of allergies is also slightly more protective than earlier age. However, the mechanism for these associations is unknown, and they do not suggest clinical actionability. Allergy association studies also suffer from recall bias, as most allergies occur in childhood, whereas PDAC occurs in adults.
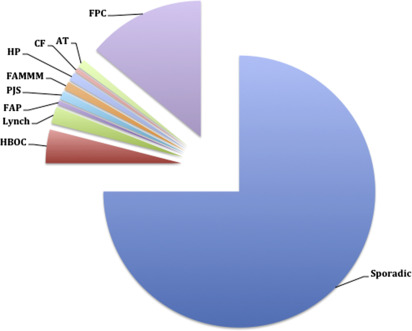
A hereditary component is implied in up to 10% of PDAC cases given the family history of an affected first-degree relative. Highly penetrant alleles in known cancer susceptibility genes account for 10% to 15% of familial cases. Characterized genetic conditions are associated with these high penetrance alleles, including the hereditary breast and ovarian cancer syndrome (HBOC), LS, familial adenomatous polyposis (FAP), Peutz-Jeghers syndrome (PJS), familial atypical multiple mole melanoma syndrome (FAMMM), hereditary pancreatitis (HP), cystic fibrosis (CF), and ataxia-telangiectasia (AT) ( Fig. 1 ). These genetic conditions have been shown to increase the risk of PDAC anywhere from 2- to 132-fold ( Table 2 ). The remaining 85% to 90% of cases with strong family histories lack mutations in these aforementioned syndromic cancer genes. These are referred to as familial pancreatic cancer (FPC). Genetic models point toward autosomal-dominant transmission of fairly high penetrant mutations. The syndromes are discussed later.
| Syndrome | Genes | RR for PDAC | Other Associated Cancers |
|---|---|---|---|
| HBOC | BRCA1 | 2–3 | Breast, ovarian, prostate, testicular, melanoma |
| BRCA2 | 3.5 | ||
| PALB2 | 6 | ||
| LS | MLH1, MSH2, MSH6, PMS2, EPCAM | 8.5 | Colon, endometrium, ovary, stomach, small intestine, urinary tract, brain, cutaneous sebaceous glands |
| FAP | APC | 4.5 | Colon, desmoid, duodenum, thyroid, brain, ampullary, hepatoblastoma |
| PJ | STK11/LKB1 | 130 | Esophagus, stomach, small intestine, colon, lung, breast, uterus, ovary |
| FAMMM | CDKN2A | 13–65 | Melanoma |
| HP | PRSS1, SPINK1 | 50–70 | NA |
| CF | CFTR biallelic | 5.3 | NA |
| CFTR monoallelic | 1.4 | ||
| AT | ATM biallelic | NA | Leukemia, lymphoma |
| ATM monoallelic | 5 | ||
| FPC | Unknown | 1.8–32 | NA |
There are several common, low-penetrant alleles that mediate genetic risk for PDAC in the general population, identified through both genome-wide association studies (GWAS) and candidate gene studies. There have been 8 GWAS for PDAC that have identified 43 alleles mapped to 35 genes associated with PDAC risk at genome-wide significance, the ORs of which range from 1.19 (95% CI 1.11–1.27) to 3.73 (95% CI 2.24–6.21). The populations studied include those of European, Chinese, and Japanese ancestry. The most interesting finding has been the rs505922 SNP, an intron in the ABO gene on chromosome 9, with an alternate allele frequency of 0.35. Carriers have an OR of 1.2 (95% CI 1.12–1.28) for PDAC. Subsequent studies have genotyped blood groups in large numbers of cases and controls demonstrating that, compared with blood type O, the ORs for PDAC in subjects with types A, AB, and B blood types were 1.38 (95% CI 1.18–1.62), 1.47 (95% CI 1.07–2.02), and 1.53 (95% CI 1.21–1.92), respectively. The population attributable fraction for non-O blood type is 19.5%, remarkably. The mechanism for this association is unknown, though, and does not seem to involve chronic pancreatitis.
Based on epidemiologic studies on environmental and dietary risk factors for PDAC, several candidate gene studies have sequenced polymorphisms in genes involved in carcinogen metabolism, insulin signaling, inflammation, DNA repair, and metabolism of alcohol, folate, and Vitamin D, but these have not yielded consistent results of alleles associated with PDAC risk and outcome. A thorough review of these is beyond the scope of this article, but a good resource is found in the references.
Pathology
Greater than 90% of invasive tumors of the pancreas arise from the exocrine component of the gland. These invasive tumors comprise many different histologic types, including typical PDAC, serous cystadenocarcinoma, acinar cell carcinoma, pancreatoblastoma, solid pseudopapillary carcinoma, and many rarer types. Of these, PDAC is the most common, comprising more than 80% of cases, and the one which is discussed further here.
The anatomic site of disease is relevant to management, and tumors are divided into those arising in the head and uncinate process (ie, to the right of the superior mesenteric vein), those arising in the body (ie, between the superior mesenteric vein and the aorta), and those arising in the tail of the pancreas (between the aorta and the hilum of the spleen).
Histologic grade is assigned based on the extent of glandular differentiation, with grade 1 composed of greater than 95% glands, grade 2 composed of 50% to 95% glands, grade 3 composed of 49% or less glands, and grade 4 only in rare, tiny glandular foci. Separate groups have shown that higher grades are prognostic for worse outcomes, although the definitions used to grade tumors were not consistent across their studies.
Pathologic staging is most commonly done according to the AJCC TNM. The T stage is based on tumor size and on involvement of the arterial celiac axis, because both are prognostic factors. The N stage is based only on the presence or absence of regional lymph node metastases, as these are prognostic factors, although most studies have shown that the absolute number and the ratio of positive to total lymph nodes are more informative for outcome than a dichotomous N stage. The M stage is based on distant metastases, including peritoneal seeding and malignant ascitic fluid. The aggregate of T, N, and M scores determines overall stage.
Pancreatic intraepithelial neoplasias (PanIN) are noninvasive, dysplastic lesions often present in PDAC resection specimens that sometimes blend with the tumor, and hence, they are presumed to be precursor lesions. They have been classed into types 1A, 1B, 2, and 3 (ie, carcinoma in situ) according to their degree of dysplasia and papillary architecture and are presumed to progress linearly through these stages from normal epithelium to invasive malignancy. There is evidence that worsening PanIN morphology is associated with increasing numbers of somatic mutations in cancer-associated genes in the PanIN itself. Two other precursor lesions for PDAC have also been proposed, namely, intraductal papillary mucinous neoplasms (IPMN) and mucinous cystadenomas (MCA), which presumably progress to invasive disease by distinct pathways from PanINs, the details of which are beyond the scope of this review.
PDAC arising in HPC syndrome carriers may be associated with distinct pathologic abnormalities.
Two series have shown that resected PDAC in FPC cases have greater numbers of PanIN lesions relative to sporadic cases, particularly PanIN 3.
No unique histologic features are strongly associated with PDAC in the setting of HBOC. Cases of acinar cell carcinoma have been reported in BRCA1 variant carriers.
PDAC in LS patients may have characteristic histologic features. Case series have shown that PDAC with microsatellite instability are more often poorly differentiated with expanding borders and a syncytial growth pattern, so-called medullary phenotype by some authors. In contrast with microsatellite unstable colorectal cancers, neither Crohn-like lymphoid infiltrate nor extracellular mucin production is prominent in these PDAC. Molecular descriptions of microsatellite unstable PDAC compared with sporadic tumors are lacking, although they have been noted in small series to be KRAS wild type with diploid genomes. Not all PDAC with medullary histology are microsatellite unstable, however, and, due to their rarity, the yield of germline testing for germline mismatch repair genes in a large sample of medullary cases is unknown. Also, cases of acinar cell carcinoma have been reported in LS patients as well.
PDAC in PJS and FAP patients may arise via IPMN precursors rather than via PanIN precursors. There are sparse clinical and molecular reports that suggest an association between PJS and IPMN and FAP and IPMN, including some by the authors’ institution, but this has unfortunately not been studied in large PJS or FAP cohorts due to the compounded rarities of PJS, FAP, and IPMN. Targeted sequencing of IPMN specimens has not revealed a high frequency of STK11 or APC somatic mutations.
Hereditary syndromes associated with pancreatic ductal adenocarcinoma
Overall
Most studies find familial clustering in up to 10% of PDAC cases, defined as having 2 affected first-degree relatives, although multicenter, population-based series that considered only medically proven PDAC diagnoses have shown familial clustering in only 1% to 3% and a meta-analysis of 9 studies places the population attributable fraction at approximately 1%. In up to 15% of these, a known cancer susceptibility gene cosegregates with the disease, and those are reviewed here (see Fig. 1 ), collectively referred to as HPC syndromes. The cause in the remaining 85% to 90% of families is unknown, and these are referred to as FPC. The clustering in these families may be due to shared genetic factors, common environmental factors, or random chance, although an inherited genetic factor is thought to be the primary cause based on family, twin, case-control, and cohort studies. Segregation analysis of 287 PDAC families estimated that 3 in every 500 individuals carry a predisposing germline allele with high penetrance for PDAC, with corresponding 32% risk of PDAC by age 85.
Most studies of the prevalence, penetrance, and clinicopathologic features of HPC syndrome germline variants in PDAC focus on specific subsets of PDAC patients, based on ethnicity or particular family histories, and consider only one or several syndromes given the limits of sample size and cost of genetic testing. It is from such studies that nearly all of the information presented herein is derived, summarized in Table 2 .
With the maturity of high-risk registries, biorepositories, and next-generation sequencing technologies, multigene testing by targeted panels of large PDAC cohorts is now possible, allowing better estimates of the role of HPC syndromes in PDAC. The authors recently published a study on PDAC using stratified random sampling to select 290 probands and found a prevalence of 3.8% (95% CI 2.1–5.6) of HPC syndrome variants in their population-based registry in the province of Ontario, Canada. Although the stratification was designed to minimize the variance estimate of BRCA1 and BRCA2 prevalence, they were surprised to find a prevalence of LS nearly as high as that of HBOC in their study.
The authors have subsequently completed BRCA testing on an unselected, consecutively, and prospectively collected cohort of 306 clinic-based patients showing that 4.6% of PDAC patients have pathogenic BRCA1 or BRCA2 germline variants.
As the current knowledge of each syndrome is reviewed, it is worth emphasizing that with increased adoption of next-generation sequencing by clinical laboratories and increased recognition of the value of genetic testing for those with cancer diagnoses, the currently accepted, well-established phenotypes of hereditary cancer syndromes described in later discussion will likely expand, and prevalence and penetrance estimates will change.
Hereditary Breast and Ovarian Cancer Syndrome
HBOC syndrome is caused by germline heterozygous inactivation of BRCA1, BRCA2, or PALB2 genes. These tumor suppressor genes code for proteins involved in double-stranded DNA break repair.
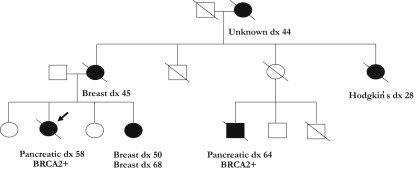
Pathogenic BRCA1 and BRCA2 alleles have a high penetrance for ovarian cancers in women (24% and 8.4% by age 80 for BRCA1 and BRCA2, respectively), breast cancers in women (90% and 41% by age 80 for BRCA1 and BRCA2, respectively), and breast cancers in men (1.2% and 6.8% for BRCA1 and BRCA2, respectively). An example of a BRCA2 pedigree is provided in Fig. 2 . Germline PALB2 variants have also been associated with increased relative risk of breast cancer.
The prevalence of pathogenic germline BRCA1 and BRCA2 variants in the general population is 1 in 400 combined, two-thirds of which is BRCA2 and one-third of which is BRCA1, while in the Ashkenazi Jewish (AJ) population, it is 1 in 40 combined. Those Ashkenazi founder mutations include the BRCA1 185delAG, the BRCA1 5382ins, and the BRCA2 6174delT.
Prevalence estimates in PDAC patients vary widely in the literature due to small sample sizes, retrospective cohorts, selective inclusion criteria (eg, by race), and limited genetic testing (eg, only specific alleles). The reported prevalence of deleterious BRCA2 germline mutations ranges from 5% to 20% in FPC kindreds and 7% in sporadic PDAC. The prevalence of the BRCA1 and BRCA2 founder mutations in a series of AJ with PDAC was found to range from 1% to 8% and 4% to 12%, respectively. As stated above, the authors found a prevalence of 4.6% for BRCA1 and BRCA2 germline pathogenic variants in 306 unselected, consecutive, clinic-based PDAC patients (1% BRCA1, 3.6% BRCA2). Although this is lower than previous estimates, it is likely more accurate given the larger sample size and unbiased study design.
BRCA1/2 are also part of the Fanconi anemia gene family, and thus, biallelic defects give rise to the recessive Fanconi anemia phenotype described in later discussion.
Some series have shown that heterozygous BRCA1 germline carriers have 2 to 3 times the risk of PDAC relative to the general population, whereas other studies have not shown an increased relative risk.
There is no disagreement in the literature that BRCA2 heterozygous germline carriers are at increased risk for PDAC, with a roughly 3.5 relative risk (range 2.3–7 across studies). Given its high prevalence, BRCA2 is the most frequent inherited risk factor for PDAC.
Age of PDAC onset has not been consistently shown to be earlier in BRCA1 and BRCA2 carriers compared with the general population.
Pathogenic germline variants in PALB2 were recently found to increase risk of PDAC, and this has been validated by some other groups but not by all. The prevalence of PALB2 in PDAC is likely quite low, as these studies have shown, estimated at less than 3% of FPC kindreds. Notably, no deleterious PALB2 variants were found in the authors’ targeted sequencing study. The relative risk of PDAC in PALB2 carriers may be as high as 6 times according to one study.
Because BRCA2 and PALB2 are Fanconi anemia genes, germline variants in other Fanconi anemia genes have been investigated in familial PDAC. The largest efforts have been in FANCA, FANCC, and FANCG. The current consensus is that there is not sufficient evidence to attribute increased PDAC risk to germline variants in these genes.
Lynch Syndrome
This syndrome is caused by heterozygous germline inactivation of any of 4 genes in the mismatch repair pathway, namely, MLH1, MSH2, MSH6, and PMS2, or by deletions of the EPCAM gene. The latter results in hypermethylation of the MSH2 promoter, silencing one copy of MSH2.
The penetrance of cancer phenotypes in LS patients varies considerably, but it is 50% to 80% for colorectal, 25% to 60% for endometrial, 5% to 15% for gastric, 5% to 10% for ovarian cancer, 3% to 6% for small bowel, 1% to 10% for dermatologic, and 1% to 4% for urothelial cancers, overall. The variable penetrance is partly dependent on which specific gene is inactivated and also partly attributed to other germline variants that act as genetic modifiers of cancer risk.
LS syndrome has a high population prevalence, estimated at 1:440. The authors’ retrospective study suggested that the prevalence of LS in PDAC was nearly as high as that of HBOC. Before the retrospective study, only 2 studies had sequenced mismatch repair (MMR) genes in PDAC cohorts sampled for ethnicity and family history, and they are thus not directly comparable.
Biallelic MMR defects lead to constitutional mismatch repair deficiency syndrome (CMMRDS). Carriers develop primary tumors of the central nervous system and hematopoetic system in childhood. No cases of PDAC have been reported in CMMRDS.
LS carriers have up to 8.5 times the relative risk of PDAC, although estimates in the literature do vary. Given their comparable relative risks and population-based prevalence, it is perhaps not surprising that the frequency of LS in the authors’ randomly sampled cohort of PDAC patients was nearly equal to that of HBOC. The relative risk for PDAC may depend on the inactivated MMR gene, as with other LS spectrum malignancies, but there is currently no evidence of this variability.
The age of onset of PDAC in LS patients has not been consistently shown to be earlier compared with sporadic PDAC cases.
Germline variants in other MMR pathway genes have neither been consistently associated with LS in general nor with PDAC specifically.
Familial Adenomatous Polyposis
This syndrome is caused by heterozygous germline inactivation of the APC gene, which is responsible for β-catenin degradation and microtubule stability.
The penetrance for colorectal cancer depends on the germline variant. The phenotype is divided into classic (∼90%) and attenuated (∼10%) depending on the number of colonic polyps detected, and the colonic phenotype is associated with germline variants in specific regions of the APC gene. In classic FAP, adenomas develop by the age of 20 and colorectal cancer by the age of 40 in 100% of carriers. In attenuated FAP, median age at CRC diagnosis is ∼50 years, and the penetrance is estimated at 70% by age 80 years. All carriers are also at risk for malignancies in extracolonic tissues, including soft tissue, duodenum, thyroid, brain, ampulla, and liver.
The estimated prevalence of FAP is 2 to 3 per 100,000.
FAP patients have roughly 4.5 times relative risk of periampullary malignancies, mostly duodenal or ampullary cancers, but also PDAC. Increased risk of PDAC in attenuated FAP has not been shown, but presumably it is similar to classic FAP as with other extracolonic features, such as desmoid tumors and periampullary neoplasia.
The age of onset of PDAC in FAP has not been shown to be earlier compared with sporadic PDAC cases.
Although approximately 25% of patients with an FAP phenotype do not have known germline variants in APC, other genes have not been found to cause FAP.
Peutz-Jeghers Syndrome
This syndrome is caused by heterozygous germline inactivation in the STK11/LKB1 gene, a serine-threonine kinase whose exact function is not well described but may have to do with mammalian target of rapamycin activity.
The penetrance for all associated malignancies is 93% by the age of 64, including esophageal, stomach, small intestine, colon, pancreas, lung, breast, uterus, and ovarian cancers, and 100% penetrance for gastrointestinal hamartomas and mucocutaneous pigmentation.
Population prevalence estimates range widely from 1:25,000 to 1:280,000. PJS occurs in all racial or ethnic groups.
PJS has a roughly 130 times relative risk of PDAC, the highest of any predisposition syndrome, although its rarity makes its frequency in a randomly sampled PDAC cohort quite low. The lifetime risk of PDAC in PJS ranges from 11% to 32%.
Pathogenic germline variants in STK11/LKB1 account for up to 80% of individuals with a clinical diagnosis of PJS. However, no other predisposing genetic locus has been identified or associated with either PJS or PJS and PDAC specifically.
Familial Atypical Multiple Mole Melanoma Syndrome
It is estimated that 40% of cases of this syndrome are caused by heterozygous germline inactivation of the CDKN2A gene, which encodes the p16 INK4A protein that functions in cell-cycle regulation.
The penetrance for malignant melanoma in p16 carriers is 60% to 90% by the age of 80 years.
The prevalence of CDKN2A in PDAC is quite low and hence poorly estimated. There is a Dutch founder mutation, named p16-Leiden, which is a 19-base-pair deletion in exon 2 of the CDKN2A gene.
The relative risk for PDAC in FAMMM carriers has been reported in small series and ranges broadly from 13- to 65-fold. Many series suggest that different CDKN2A variants have different penetrance for PDAC, explaining that only 60% develop PDAC. There are reported CDKN2A kindreds with PDAC and no history of melanoma. Those with the p16-Leiden founder allele have a 17% risk of developing PDAC by age 75. The risk of PDAC is greater in smokers.
FAMMM due to germline variants in other genes, including CDK4 and MITF, has not been associated with increased PDAC risk.
Hereditary Pancreatitis
This syndrome is caused by germline variants in PRSS1 and SPINK1 genes. PRSS1 encodes trypsinogen, and SPINK1 encodes a trypsin inhibitor. The pathogenic germline variants result in either premature activation or reduced inhibition of the digestive trypsin enzyme, leading to pancreatic injury. Pathogenic PRSS1 alleles follow an autosomal-dominant mode of inheritance, while SPINK1 alleles may follow autosomal-recessive inheritance. The exact mechanisms of action are unclear, and germline variants in these genes interact with other genetic and environmental factors to precipitate pancreatitis in ways that are outside the scope of this review.
As a result of these [epi]genetic and environmental modifiers, the penetrance of pancreatitis ranges from 40% to 90% in different studies.
The population prevalence of HP has been estimated at 3 in 1,000,000, and the frequency of PRSS1 and SPINK1 germline variants in unselected patients with chronic or idiopathic pancreatitis can be as high as 2% to 4%.
HP patients have a 50 to 70 times relative risk for PDAC compared with the general population. That risk has been shown to be as much as double in HP patients who are also current smokers.
It is estimated that 30% to 40% of HP carriers develop PDAC by age 70 years. The age of onset of PDAC in HP patients has been shown to be lower by as much as 20 years from the average in current smokers.
Other genes have been associated with HP, but an associated risk of PDAC is not as well described.
Cystic Fibrosis
This syndrome is caused by biallelic inactivating variants in the CFTR gene, which codes for a plasma membrane ion transporter. Its complete inactivation leads to thickened mucous that obstructs hollow viscera causing recurrent disease of the exocrine pancreas, intestine, respiratory tract, male genital tract, hepatobiliary system, and exocrine sweat glands. There is poor genotype-phenotype correlation, implying both modifier genes and environmental factors; these are poorly described.
More than 2000 variants in CFTR have been described, of which less than 150 are likely to be pathogenic. Most of these alleles are fully penetrant.
The carrier frequency for CFTR varies with ethnic group, being highest in Northern Europeans and AJ at roughly 1:30, resulting in a disease incidence of 1 in 3200 live births in these groups.
The relative risk of PDAC in CF is 5.3 times that of the general population. However, the prevalence of PDAC in CF is quite low given that the overall median survival in CF is only 36 years. The relative risk of PDAC in CFTR variant carriers is 1.4 times that of the general population. Other studies have failed to show an association between monoallelic CFTR variant carriers and PDAC risk.
The age of onset of PDAC is much lower in CF patients, with a median of 35 years. The age of onset in CFTR variant carriers is also lower, with a median of 62 years, or 60 years in those who are also smokers.
Ataxia Telangiectasias
This syndrome is caused by biallelic germline inactivation of the ATM gene that codes for a serine/threonine kinase involved in repair of DNA double-stranded breaks.
Biallelic inactivation is thought to be fully penetrant for the clinical phenotypes of neurologic disorders, blood vessel abnormalities, immune system dysfunction, sensitivity to ionizing radiation, and primary hematologic malignancies. Monoallelic inactivation is associated with an increased risk of malignancy, especially breast cancer, for which the penetrance in carriers of ATM protein truncating variants has been reported at 60% by age 80 years.
The prevalence of biallelic inactivation is 1 in 40,000 to 1 in 100,000 live births, and this prevalence varies with the degree of consanguinity in a country. The prevalence of monoallelic inactivation is estimated at 0.5% to 1%. The prevalence of monoallelic ATM inactivation in PDAC probands has been reported to be as high as 2.4%.
The relative risk of PDAC in monoallelic carriers is twice that of the general population. The association of PDAC with monoallelic ATM inactivation carriers was first suspected in population and family-based studies and then validated by a recent sequencing study in an FPC cohort. The high frequency of monoallelic ATM inactivation and the low frequency of PDAC in the general population imply that the PDAC risk in monoallelic carriers is modified by either environmental or other genetic factors that remain to be discovered.
Familial Pancreatic Cancer
This syndrome is defined as pedigrees with 2 or more first-degree relatives with PDAC in the absence of a known genetic cause. An example of such a pedigree is provided in Fig. 3 .
The syndrome is quite prevalent, accounting for as many as 90% of familial cases or up to 10% of all PDAC cases.
One of the largest studies showed that the relative risk of PDAC for first-degree relatives of FPC cases is 9 times greater than it is among first-degree relatives of sporadic PDAC cases. The risk is proportional to the number of first-degree relatives with PDAC, increasing with 1 (4.6; CI, 0.5–16.4), 2 (6.4; CI, 1.8–16.4), or 3 (32.0; 95% CI, 10.2–74.7) first-degree relatives with PDAC. However, a meta-analysis of 9 studies found a relative risk of 1.80 (95% CI: 1.48–2.12) regardless of the degree of relatedness, and 1.71 (1.37–2.05) when stratified for those with affected first-degree relatives, indicating that there is a range in observed risk in different studies and that shared, nongenetic factors may account for some of the risk. In fact, smoking has been shown to be an independent risk factor in FPC.
Most series have not found a significant difference in age at diagnosis. Nevertheless, some series have shown genetic anticipation of PDAC across generations of FPC pedigrees, with PDAC onset approximately 10 years earlier than in affected parents in roughly 70% of pedigrees.
Genetic modeling suggests an autosomal-dominant, high-penetrant allele responsible for the syndrome. The probability of extrapancreatic malignancies in relatives of FPC probands is greater than in relatives of sporadic PDAC probands, particularly melanoma and endometrial cancer in one study, although a personal history of malignancy is not.
Linkage analysis of a large American FPC pedigree showed significant linkage (logarithm of odds score 5.36) in the region of chromosome 4q32-34. A subsequent study identified a missense allele in the PALLD (palladin) gene as cosegregating with disease in this pedigree, but neither this linkage result nor this variant has been validated by other groups. No other candidates have been put forward since, and all available unpublished and preliminary data point toward marked genetic heterogeneity in FPC, although non-coding variants and epigenetic effects have not been explored to date.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


