Features suggestive of hereditary RCC
Family history of RCC or confirmed syndrome
Bilaterality, multifocality, early age of onset
Histology: non-clear cell with unusual features, e.g., papillary type II morphology with orangophillic nucleoli, or hybrid oncocytic neoplasms
Associated tissue manifestations
Dermatologic: skin leiomyoma, folliculofibroma, along with multiple other skin features in TSC, BHD, and Cowden’s disease
Ophthalmologic: astrocytic hamartoma (phakoma), retinal angiomas
Neurologic: hemangioblastoma, giant cell astrocytoma, cortical tubers, and subependymal nodules
Endocrine: pheochromocytoma, paraganglioma, adrenal nodules, pancreatic cysts, and neuroendocrine tumors
Gynecologic: early onset of uterine fibroids (<30)
Pulmonary: lung cysts
Aggressive surgical excision of all renal tissue is no longer the standard of care. Enucleation and partial nephrectomy have been used with success to preserve renal function in many renal cancer syndromes.
Close observation with a 3 cm threshold for surgical intervention can be the guiding principle for managing von Hippel-Lindau (VHL), hereditary papillary renal cell (HPRC), and Birt-Hogg-Dubé (BHD).
Aggressive hereditary kidney cancers such as SDH and HLRCC should not be observed; these patients should be managed with early surgical intervention with wide margin.
8.1 Introduction
In 2014, there are over 65,000 newly diagnosed cases of kidney cancer, responsible for nearly 14,000 deaths [1]. While the death rates fell by 0.9 % per year from 2006 to 2010 [1], it is still very much a lethal disease. Current expert opinion suggests that while the overwhelming majority of RCC is sporadic in nature, hereditary kidney cancer accounts for 5–8 % of cases [2–4]. This number may be a gross estimation and with the explosion of next-generation sequencing techniques. More syndromes are expected to be discovered, some with highly complex inheritance patterns.
Currently, there are more than a dozen confirmed types of hereditary kidney cancer syndromes. Many share overlapping dysregulation in metabolic pathways involved in the cells ability to respond to changes in nutrients, oxygen sensing, and iron metabolism [5]. While there are shared dysregulation in similar pathways, their degree of renal penetrance and aggressiveness vary in addition to the types of extrarenal manifestations. The more prominent hereditary RCCs will be discussed in the following chapter, along with a summary of their management.
8.2 Clinical Recognition
Management of hereditary RCC may have substantial differences from that of its sporadic counterpart, and therefore, recognition is critical. While clinicians should avoid screening every patient with a renal mass for genetic anomalies, a certain pattern of clinical presentation and family history should trigger further workup (Table 8.1). First and foremost, a positive family history of diagnosed syndrome or bilateral/multifocal renal lesions should prompt further investigation, even if the patient in question is noted to have benign neoplasm of the kidney. Age of onset of ≤46 years of age is another criterion, derived from a study based on the SEER database along with the National Cancer Institute (NCI) experience. It was found that 70 % of hereditary kidney cancer fell below the bottom decile in age, and referral to genetic counselors based on this threshold could maximize sensitivity and specificity for detection of a hereditary form of kidney cancer [4]. Additional features foretelling of hereditary RCC include non-clear cell with unusual histologic features [6] and associated physical manifestations (i.e., dermatologic, gastrointestinal, ophthalmologic, neurologic, endocrine, gynecologic, and pulmonary). As such, Reaume and colleagues published a clinical practice guideline aimed to guide clinicians in identifying patients with hereditary RCC [6].
8.3 Syndrome Manifestations
8.3.1 Von Hippel-Lindau
Von Hippel-Lindau (VHL) disease was the first heritable RCC to be discovered. Its characterization in the early twentieth century led to subsequent breakthroughs applicable to even the sporadic form of the disease, where over 90 % of cases are found to harbor the similar genetic alterations [7]. Determination of the responsible gene by the NCI began with the observation that there was a consistent loss of chromosome 3p in affected individuals [8], but it was not until sometime later that the researchers localized the locus to 3p25.1 [9]. With an autosomal dominant pattern of inheritance, VHL has a high degree of penetrance. The VHL gene functions as a classic tumor suppressor [10] and requires a “second hit” for loss of the wild-type allele. The VHL gene encodes for a protein, VHL, that modulates the activities of hypoxic-induced factor (HIF), most notably HIF-1a and HIF-2a. When VHL is either absent or nonfunctional, the HIFs are stabilized even under normoxic conditions, resulting in unchecked shift toward a pseudo-hypoxic state with upregulation of anaerobic metabolism, angiogenesis, and carcinogenesis.
The clinical manifestations of VHL include hemangioblastomas (of the retina, brain, and spinal cord), pheochromocytomas, pancreatic cysts or neuroendocrine tumors (Fig. 8.1), cystadenomas of the epididymis or broad ligament, and bilateral, multifocal kidney cysts or tumors [11]. While renal cysts in non-syndromic patients are predominantly benign, individuals with VHL tend to have cysts lined with malignant cells, and there may be other microscopic foci of RCC [12]. An estimated 70 % of affected individuals develop RCC by the age of 60. Not only do these people present with multiple lesions, their age of onset was also noted to be an estimated 25 years earlier than sporadic RCC [13].
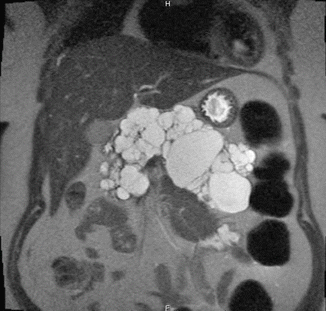
Fig. 8.1
Coronal T2-weighted MRI image of a VHL patient in whom the pancreas was completely replaced with cysts
VHL has also been divided into several subtypes based on the presence of pheochromocytoma, RCC, and type of genetic mutation. Type 1 VHL typically presents with RCC but low risk for pheochromocytomas, and its genetic alterations are germline deletions or truncating mutations. Type 2 VHL carries a low risk for RCC but the presence of pheochromocytoma is a characteristic; this type or subtype of VHL has underlying missense mutation of the affected gene [14, 15]. While useful to help understand which patients have the highest risk of each condition, clinically overlap has been observed.
8.3.2 Hereditary Papillary Renal Cancer
Early in the 1990s, Zbar et al. discovered multiple families affected by papillary renal cell carcinoma (pRCC) with a very high penetrance [16, 17]. Interestingly, the affected individuals displayed no loss of chromosome 3p, which lead the researchers to suspect a separate heritable RCC syndrome, hereditary papillary RCC (HPRC). This second hereditary RCC syndrome was later linked to 7q31 and found to be caused by a genetic mutation in the MET proto-oncogene [18]. MET encodes for a crucial tyrosine kinase receptor binding the hepatocyte growth factor (HGF), and the mutant variant demonstrates a high degree of phosphorylation corresponding to elevated enzymatic activity [19]. Activated MET signaling is believed insufficient in tumorigenesis alone, but nonrandom duplication of copies of the affected gene is commonly detected in HPRC perhaps contributing to carcinogenesis [20].
Unlike VHL, HPRC presents only with kidney tumors without any extrarenal manifestations. Without a set of distinguishing features, diagnosis can be difficult and may rely on a meticulous documentation of familial pedigree. The penetrance for this disorder is considerable. Moreover, the risk of RCC only increases with age, upward to 67 % by age 60 [21, 22]. Generally speaking, the disease is detected after the age of 30, but subtypes with earlier onset have been described [23]. HPRC presents with papillary type 1 [24], and it is not uncommon for there to be several thousand small papillary foci to be present in the renal parenchyma of affected individuals [25].
8.3.3 Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC)
Reed syndrome was first described in 1958 by dermatologists who treated a group of patients with distinct cutaneous leiomyomas [26]. It was not until sometime later that clinicians discovered that many patients had an inherited form of both cutaneous and visceral leiomyomatosis as well as an aggressive form of RCC (Fig. 8.2), leading to the syndrome being renamed hereditary leiomyomatosis and renal cell carcinoma (HLRCC) [27–29]. In this autosomal dominant disease, genetic linkage studies localized the affected gene to 1p42.3-43 that encodes for fumarate hydratase (FH) [30, 31]. This enzyme is an integral part of the Krebs cycle, catalyzing a hydration reaction of fumarate to malate. HLRCC renal tumors demonstrate a loss of heterozygosity of the wild-type FH allele, shifting cellular metabolism toward aerobic glycolysis (the Warburg effect) [32, 33].
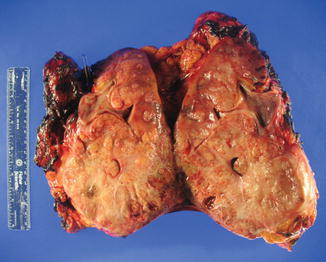
Fig. 8.2
Radical nephrectomy specimen of a 15 cm left renal mass. The tumor was believed localized to the lower pole of the kidney, but on final pathology, the tumor infiltrated throughout in all regions of the kidney
HLRCC presents with a unique constellation of signs and symptoms: leiomyoma (80 %) (Fig. 8.3), early-onset uterine fibroids in women (90 %), aggressive RCC (20 %), and, less frequently, macronodular adrenal hyperplasia (8 %) [34–36]. Initial descriptions of the associated RCC were papillary type 2, but HLRCC can present with a variety of different morphologic features. A common theme in their pathologic appearance includes eosinophilic nucleoli and perinuclear halos [33]. The International Society of Urologic Pathology now considers HLRCC renal tumors to be a distinct subtype [37].
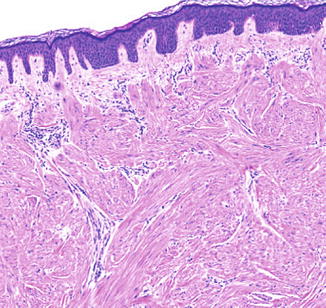
Fig. 8.3
5× magnification of a cutaneous shave biopsy of a cutaneous leiomyoma in a patient with HLRCC. The underlying dermis contains interlacing smooth muscle bundles and fascicles
8.3.4 Birt-Hogg-Dube (BHD)
BHD was discovered by a group of Canadian dermatologists in 1977. Their study of an extended family revealed 15 out of 37 members after the age of 25 developing fibrofolliculomas and associated trichodiscomas or acrochordon (Fig. 8.4) [38]. The fibrofolliculomas were extension of epithelial strands beyond the infundibulum of the hair follicles. The incidence of BHD was noted to be 1 in 200,000 and transmitted in an autosomal dominant fashion. Further linkage analysis confirmed the gene location to 17p11.2 [39], which encodes for folliculin (FLCN). The gene in question behaves like a classic tumor suppressor and was found to participate in a complex that regulates downstream functions of AMPK and mTOR [40, 41]. Hasumi and colleagues determined loss of FLCN-upregulated activities of mTORC1 and mTORC2 [42].
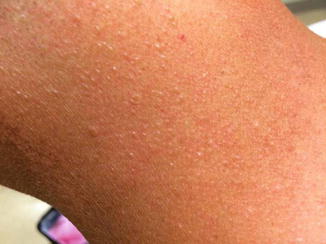
Fig. 8.4
Image of left neck of an individual with BHD demonstrating multiple small fibrofolliculomas
In addition to the skin manifestations that are pathognomonic to BHD, the affected individuals also have a greater than 50-fold increase risk in lifetime pneumothoraces, a likely sequela of their propensity in developing lung cysts [43]. After adjusting for age, those with BHD were about seven times more likely to develop renal tumors [43]. With an average onset of age 50, renal lesions in the setting of BHD tend to be bilateral and multifocal in their occurrence [44–46]. The most common tumor in BHD is a hybrid oncocytic neoplasm, which contains both chromophobes with oncocytic features [47]. These tumors tend to have better prognosis due to their relatively indolent progression: less than 5 % of affected individuals go on to develop metastatic disease [48]. Clear cell and papillary histologic subtypes are found in less than 10 % of tumors, but when present, they portend a significantly worse prognosis.
8.3.5 Succinate Dehydrogenase B/C/D
Hereditary paraganglioma and pheochromocytoma syndromes were a group of syndromes closely associated with the development of chromaffin cell tumors, namely, pheochromocytomas and, their extra-adrenal counterparts, paragangliomas (Fig. 8.5). It was initially postulated that only 10 % of such tumors possessed a genetic component, but recent gene studies have confirmed that as high as 35 % of these neoplasms without a definable syndrome are caused by a hereditary predisposition [49]. Since 2000, it became recognized that many of these syndromes were related to an alteration in a common enzyme succinate dehydrogenase; hence, the syndromes are considered to have succinate dehydrogenase deficiency (SDH). There are multiple constituents to the SDH enzyme complex that function together as an important enzyme in the Krebs cycle, converting succinate to fumarate. Located in the inner mitochondrial membrane adjacent to the matrix, SDH not only participates in the Krebs cycle but also as complex II in the electron transport chain. Its dysregulation directly results in hypoxic metabolism and aggressive neoplastic growth consistent with the Warburg effect [50]. Pertaining to hereditary kidney cancers, however, alterations in only SDH subunits B/C/D have been implicated. This association was first characterized in 2004, and it was later found that 5 % of familial RCC without a diagnosed syndrome could be mapped to SDHB germline mutation [51, 52]. A germline mutation in genes coding for the other subunits SDHC and SDHD has also been linked to an increased risk of RCC. Similar to HLRCC, kidney tumors in the setting of SDH tend to follow an aggressive course. In addition to chromaffin and kidney tumors, recent findings suggest that SDH mutations may also increase the risk of gastrointestinal stromal tumors [53].
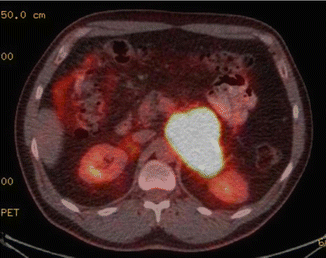
Fig. 8.5
A PET/CT imaging from a 9 cm left paraganglioma above the kidney and adjacent to the superior mesenteric artery. The patient had anxiety and intermittent mood swings due to massively elevated catecholamines. The family history was significant for a GI stromal tumor, and he was found with an SDHB alteration
8.3.6 Tuberous Sclerosis 1 and 2 (TSC1, TSC2)
The tuberous sclerosis complex is an autosomal dominant condition well known to the dermatologic and neurologic communities. Its underlying genetic mutations were successfully linked to TSC1 (9q34), hamartin, and TSC2 (16p13), tuberin [54, 55]. Similar in its signaling pathway to VHL, the loss of TSC2 in animal models has shown to upregulate HIF and mTORC1 [56, 57]. While hamartin and tuberin represent the most recognized germline mutations, the genetic basis for the renal pathologies in the setting of TSC remains ill defined.
The hallmark of tuberous sclerosis is its dermatologic manifestations. Nearly all of the affected individuals demonstrate some form of skin lesions: ash-leaf spots (hypopigmented macules), angiofibromas (also known as fibroadenomas) (Fig. 8.6), and shagreen patches found most commonly on the lower trunk. A significant portion of these people also have intracranial lesions, with the most well-known lesions being the glioneuronal hamartomas, or tubers. Moreover, they may also harbor subependymal giant cell astrocytomas (SEGAs). Surprisingly, the degree of cognitive dysfunction and epileptic risk are only loosely correlated with the burden of brain lesions.
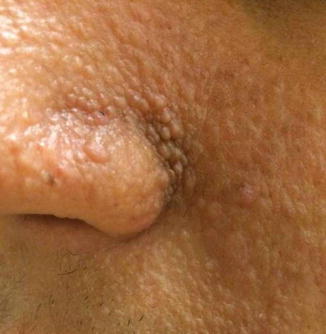
Fig. 8.6
Image of the nasal labial fold of a young man with TSC showing extensive angiofibromas/fibroadenomas
Renal manifestations of TSC tend to be highly penetrant. The common lesions include angiomyolipomas, renal cysts, and less commonly RCC, which can be of any subtype [58]. Despite the benign nature of AMLs, mass effects are common, and the clinician should always be wary of the risk of hemorrhage for lesions greater than 4 cm. It is worthy of noting that some patients with TSC suffer from chronic kidney disease, which may be the result of renin-dependent hypertension or direct parenchymal compression. While RCC arise in <5 % of patients with TSC, the lesions tend to have an early age of onset and a more aggressive clinical course [59].
8.3.7 Cowden’s Disease/PTEN
Cowden’s disease is an autosomal dominant disease thought to affect 1 in 200,000 persons, an estimate now questioned to be an underestimation [60]. The gene in question was localized to 10q22, also known as PTEN, which serves as a tumor suppressor [61, 62]. Cowden’s disease (CD) presents with a host of cutaneous and mucocutaneous lesions. The hallmarks of CD are the benign hair follicle tumors termed trichilemmomas [63]. CNS pathologies are also common, where the affected individuals manifest macrocephaly and cerebellar white matter disorganization (Lhermitte-Duclos disease). They also demonstrate symptoms of ataxia, tremors, and cognitive dysfunction. From an oncologic standpoint, CD patients tend to develop epithelial neoplasms, including malignancies of the breast, uterus, thyroid, colon, and prostate. With regard to renal neoplasms, individuals with CD demonstrate 4 % incidence of developing RCC, which is approximately 30× the lifetime risk of normal individuals [64]. In a series of patients with CD, 16 % had a history of kidney cancer involving all major histologic subtypes [65]. As such, patients with confirmed CD may benefit from early screening of kidney cancers.
8.3.8 Other Syndromes
There are a handful of lesser-known heritable syndromes with renal neoplasms still worthy of mention. First and foremost, Cohen and colleagues reported a family with hereditary kidney cancer similar to VHL notable for a balanced translocation of chromosome 3. The affected did not possess the classical extrarenal manifestations of VHL and tended to develop RCC later in life. In the ensuring decades, further population studies were conducted, namely, one involving the Danish cytogenetic and cancer registry [66]. This phenomenon is believed explained by a three-hit model of carcinogenesis: first an individual is born with an abnormal karyotype involving translocation of 3p, second there is a loss of the 3p fusion chromosome, and finally the remaining VHL allele undergoes a somatic mutation [67, 68].
Oncocytomas are the most commonly resected benign tumors of the kidney. While oncocytomas lack the invasive potential RCC, they can nevertheless cause symptoms secondary to mass effect. Approximately 10 % of oncocytomas are bilateral [69–71], and patients with bilateral lesions should be screened for BHD. In the cases of bilateral oncocytoma with negative screening for BHD, a diagnosis of bilateral multifocal oncocytomas is rendered. By the same token, a familial form of bilateral and multifocal oncocytomas has been coined familial renal oncocytoma (FRO) [72]. Patients with this condition should be observed closely, and prior to additional intervention, there should be consideration of renal biopsy. Lastly, there exists a variant form termed renal oncocytosis, where diffuse oncocytic nodules arise in the renal parenchyma, and many patients progress to renal failure [73]. The goal of management for these patients is centered on the maintenance of renal function through conservative measures.
8.4 Management
8.4.1 General Considerations
The management of hereditary renal carcinomas, though far from perfect, is not a terribly complex algorithm. Traditionally, bilateral nephrectomy was considered for patients with hereditary RCC. Such an approach predestined all of these patients to a lifelong dependence on dialysis. Some of the more fortunate individuals, who are free of recurrence, became candidates for renal transplantation [74, 75]. Current practice has departed from the paradigm of removing all renal tissue; instead, preservation of nephrons has become a principal objective. For VHL, BHD, HPRC, and familial renal cancer of unknown cancer (FRC), the 3 cm threshold for surgical intervention has been implemented with success. Utilizing systematic resection of tumors in a stepwise fashion, from most accessible to the most challenging, the surgeon and anesthesiologist maximize hemodynamic stability for the longest duration possible. Enucleation, taking no additional margin of benign tissue, has also become the preferred technique in several forms of hereditary RCC. It has been utilized successfully with good outcome in VHL, BHD, and even HPRC [76, 77]. On the other hand, patients with the more aggressive subtypes of hereditary RCC (HLRCC and SDH) demonstrate greater propensity locoregional dissemination and thereby would benefit from a more aggressive surgical approach.
Related posts:
 Genetic Heterogeneity of Kidney Cancer
Angiogenesis Inhibitor Therapy in Renal Cell Cancer
Presurgical Therapy in Renal Cell Carcinoma
Genetic Heterogeneity of Kidney Cancer
Angiogenesis Inhibitor Therapy in Renal Cell Cancer
Presurgical Therapy in Renal Cell Carcinoma
 Cytokines in the Management of Advanced Renal Cell Cancer
Cytokines in the Management of Advanced Renal Cell Cancer
 Toxicity Management of Renal Cell Cancer Patients on Targeted Therapies
Toxicity Management of Renal Cell Cancer Patients on Targeted Therapies
 Renal Cell Carcinoma: Clinical Presentation, Staging, and Prognostic Factors
Renal Cell Carcinoma: Clinical Presentation, Staging, and Prognostic Factors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


