uncertain significance, where a single-nucleotide change predicts an amino acid substitution that is not certain to sufficiently modify protein function to cause disease. Such “missense” changes have the potential to lead to misdiagnosis, as evidenced by families in which the alternate “real” mutation is also identified.13,14
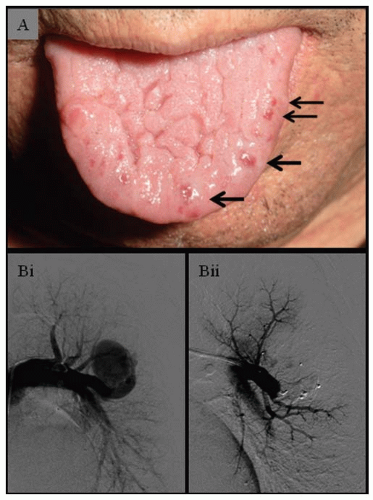 FIGURE 70.1 Clinical HHT. A: Tongue demonstrating typical florid telangiectasia. Note that these lesions increase with age and are often subtle or absent in younger individuals. B(i): Selective left pulmonary angiograms demonstrating large left lower lobe PAVM pretreatment, when it was associated with severe hypoxemia (SaO2 85% at rest) and normal pulmonary artery pressure (27/9, mean 16 mm Hg). B(ii): Following embolization of separate feeding arteries using four Amplatzer plugs, the SaO2 rose to 95% at rest, but the pulmonary artery pressure was unchanged (28/8, mean 15 mm Hg). Angiograms courtesy of Dr James Jackson. |
New data suggest that where screening investigations have already identified abnormal liver function tests in an HHT family, Doppler studies may also be of value to predict a group with more severe disease who may require different follow-up regimes.18
Table 70.1 HHT diagnostic criteria | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
an HHT center, allowing for genetic counseling of the families, coordinated with patient care by expert physicians.
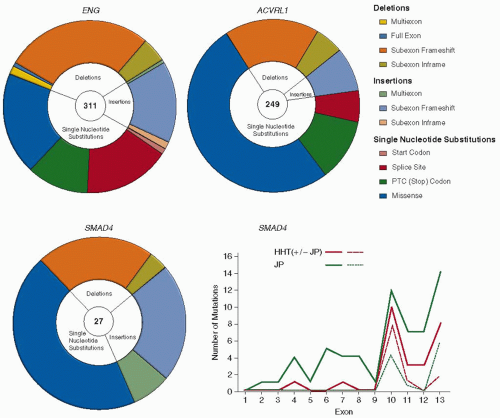 FIGURE 70.2 HHT mutations. Distribution of different types of mutations in ENG, ACVRL1, and SMAD4 genes. PTC: premature termination codon generated directly, or by frameshift mutations. Note the higher proportion of missense single-nucleotide substitutions in ACVRL1. The graph illustrates the distribution of SMAD4 mutations reported for HHT patients (red lines) and JP alone (green lines). Whereas for ENG and ACVRL1, mutations occur throughout the respective genes,96 note the limited and highly comparable SMAD4 mutational distribution for the two pathologies, for all mutations (solid lines), and missense substitutions affecting exons 10 to 13 (dotted lines). (Data derived from hht.mutation.org and Wooderchak WL, Zacary S, Crockett DK, et al. Repository of SMAD4 mutations: reference for genotype/phenotype correlation. J Data Mining Genom Proteomics 2010;1:101.) Ref.126. |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


