INTRODUCTION
SUMMARY
Hereditary fibrinogen abnormalities make up two classes of plasma fibrinogen defects: (1) type I, afibrinogenemia or hypofibrinogenemia, in which there are low or absent plasma fibrinogen antigen levels (quantitative fibrinogen deficiencies), and (2) type II, dysfibrinogenemia or hypodysfibrinogenemia, in which there are normal or reduced antigen levels associated with disproportionately low functional activity (qualitative fibrinogen deficiencies).* In afibrinogenemia, most mutations of the three encoding genes of fibrinogen chains are null. In some cases, missense or late-truncating nonsense mutations allow synthesis of the corresponding fibrinogen chain, but intracellular fibrinogen assembly and/or secretion is impaired. In certain hypofibrinogenemic cases, the mutant fibrinogen molecules are produced and retained in the rough endoplasmic reticulum of hepatocytes in the form of inclusion bodies, causing endoplasmic reticulum storage disease. Afibrinogenemia is associated with mild to severe bleeding, whereas hypofibrinogenemia is often asymptomatic. Thromboembolism may also occur and affected women may suffer from recurrent pregnancy loss. Hereditary dysfibrinogenemias are characterized by biosynthesis of a structurally abnormal fibrinogen molecule that exhibits reduced functional properties. Dysfibrinogenemia is commonly associated with bleeding, thrombosis, or both thrombosis and bleeding, but in many patients it is asymptomatic. Hypodysfibrinogenemia is a subcategory of this disorder. Certain mutations involving the C-terminus of the fibrinogen α chain are associated with amyloidosis, in which an abnormal fragment from the fibrinogen α C domain is deposited in the kidneys. The cause for thrombophilia in type II fibrinogen abnormalities often is uncertain but may involve defective calcium binding, impaired tissue-type plasminogen activator-mediated fibrinolysis, resistance to fibrinolysis, or reduced thrombin binding to fibrin. Replacement therapy with fibrinogen concentrates has proven to be useful for management of fibrinogen disorders but should be adapted to each patient, based on the personal and family history.
Acronyms and Abbreviations
FFP, fresh-frozen plasma; FGA, fibrinogen Aα-chain gene; FGB, fibrinogen Bβ-chain gene; FGG, fibrinogen γ-chain gene; FpA, fibrinopeptide A; FpB, fibrinopeptide B; LMWH, low-molecular-weight heparin; PCR: polymerase chain reaction; TAFI, thrombin-activatable fibrinolysis inhibitor; t-PA, tissue-type plasminogen activator.
Several detailed and thoroughly annotated reviews of mutations causing inherited fibrinogen disorders have been published1,2,3 and the previous version of this chapter published in the eighth edition4 contained tables compiling causative mutations identified before 2009. In addition, a registry for hereditary fibrinogen abnormalities5 can be accessed at http://www.geht.org/databaseang/fibrinogen/ that lists variants reported in publications, conference abstracts, and submitted online, with original references. This chapter discusses the major molecular mechanisms leading to disease, as well as the laboratory and clinical aspects of fibrinogen disorders and their treatment, without listing all fibrinogen gene anomalies.
Fibrinogen plays a major role in hemostasis as the precursor molecule for the insoluble fibrin clot (Fig. 125–1). In addition fibrinogen participates in numerous other biologic processes, such as inflammation, wound healing, and angiogenesis. Fibrinogen binds plasminogen, α-antiplasmin, fibronectin, and factor XIII, among other proteins. It also binds to platelets and supports platelet aggregation. After fibrinogen is converted to fibrin by thrombin, it provides nonsubstrate binding sites for thrombin; consequently, fibrinogen is sometimes termed antithrombin I.6 Fibrinogen also binds to vascular endothelial and other cells, plasma or tissue matrix components such as fibronectin and glycosaminoglycans, and peptide growth factors. Fibrin provides a template for assembly and activation of the fibrinolytic system components and is the major substrate for the enzyme plasmin (Chap. 135). Both fibrinogen and fibrin serve as substrates for plasma factor XIIIa that catalyzes covalent crosslinking/ligation.

*The authors thank Dr. Alessandro Casini for helpful comments and suggestions.
STRUCTURE AND SYNTHESIS
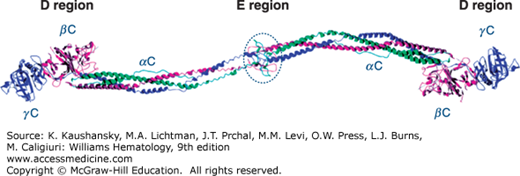
Fibrinogen is a 340-kDa glycoprotein synthesized in hepatocytes7 that circulates in plasma at a concentration of 1.5 to 3.5 mg/mL (~4 to 10 μM). Each fibrinogen molecule is approximately 45 nm in length. The core structure consists of two outer D regions (or D domains) and a central E region (or E domain) connected through coiled-coil connectors (Fig. 125–2).8 The molecule exhibits a twofold axis of symmetry perpendicular to the long axis, consisting of two sets of three polypeptide chains (Aα, Bβ, γ) that are joined in their aminoterminal regions by disulfide bridges to form the E region. The outer D regions contain the globular C terminal domains of the Bβ chain (βC) and γ chain (γC). The βC and γC domains, which are highly conserved in vertebrates, are members of the FreD (fibrinogen-related domain) family of proteins. Unlike the βC and γC domains, the C-terminal domains of the Aα chain (αC) are intrinsically unfolded and flexible and tend to be noncovalently tethered in the vicinity of the central E region (Fig. 125–2). The three genes encoding fibrinogen Bβ (FGB), Aα (FGA), and γ (FGG), ordered from centromere to telomere, are clustered in a region of approximately 50 kb on human chromosome 4.9 FGA and FGG are transcribed from the reverse strand, in the opposite direction to FGB. Alternative splicing10 results in two isoforms for the fibrinogen α chain: the common Aα chain, encoded by exons 1 to 5, and an extended Aα-E isoform, encoded by exons 1 to 6 which represents only 1 to 2 percent of transcripts. Alternative splicing for FGG also produces two transcripts: the major mRNA species contains all 10 exons and encodes the common γ chain (or γA), while the minor product (γ′) does not splice out intron 9 and the corresponding open reading frame replaces the four codons of exon 10 with 20 alternative codons. FGB encodes a single 1.9-kb transcript with a 1.5-kb coding sequence. Each gene is separately transcribed and translated to produce nascent polypeptides of 644 amino acids (Aα), 491 amino acids (Bβ), and 437 amino acids (γ).
Figure 125–2.
Ribbon representation of native chicken fibrinogen22 modified from PDB file 1M1J (www.pdb.org/pdb/). α Chains are in green, β chains are in purple, and γ chains are in blue. The globular C-terminal domains of the Bβ and γ chains forming the D regions are shown, as well as the central E region, which contains the N-terminal portions of all three chains. Unlike the βC and γC domains, the C-terminal domains of the Aα chain (αC) are flexible and tend to be noncovalently tethered in the vicinity of the central E region.
During translocation of the single chains into the lumen of the endoplasmic reticulum (ER), a signal peptide is cotranslationally cleaved from each chain. The resulting chains have 625 amino acids (Aα), 461 amino acids (Bβ), and 411 amino acids (γ). Assembly proceeds in the ER with the formation of an Aα-γ or Bβ-γ intermediate. The addition of either a Bβ or Aα chain gives rise to a [AαBβγ] half-molecule, which dimerizes to form the functional hexamer.11 The protein undergoes several posttranslational modifications in the Golgi complex, including maturation of N-linked oligosaccharides, phosphorylation, hydroxylation, and sulfation.12
Following assembly, which is completed within minutes, the mature molecule is constitutively secreted into the circulation, where it exhibits a half-life of approximately 4 days.13 In addition to plasma fibrinogen, blood contains an internalized intracellular fibrinogen pool that is stored within platelet α granules. Both megakaryocytes and platelets are capable of internalizing plasma fibrinogen via the fibrinogen integrin αIIbβ3 receptor,14 which binds to a C-terminal platelet recognition sequence that is present on γA chains but is absent from γ′ chains. Consequently, internalized platelet fibrinogen molecules contain only γA chains.15
FIBRINOGEN CONVERSION TO FIBRIN AND NETWORK ASSEMBLY
Fibrin polymerization consists of several consecutive reactions, each affecting the ultimate structure and properties of the fibrin scaffold, which, in turn, determines the development and outcome of numerous diseases including coagulopathies and thrombosis.16,17 Conversion of fibrinogen to a fibrin clot18 occurs in three distinct phases: (1) enzymatic cleavage by thrombin to produce fibrin monomers; (2) self-assembly of fibrin units to form an organized polymeric structure; and (3) covalent crosslinking of fibrin by factor XIIIa. In the first phase of conversion to fibrin, cleavage of fibrinogen at AαR35/G36 (R16/G17)* and later Bβ R44/G45 (R14/G15) results in release of fibrinopeptides A (FpA) and B (FpB), respectively, thus exposing “A” knobs and “B” knobs (Fig. 125–3). The “A” knob located at the new aminoterminal end of the fibrin α chain starts with the GPRV amino acid sequence. The “A” knob in fibrin interacts with the constitutive complementary association site known as hole “a” in another molecule to initiate the fibrin assembly process. Hole “a” is encompassed by residues 363 to 405 (337 to 379) of the γ chain.
Figure 125–3.
First steps of fibrinogen conversion to fibrin and fibrin assembly. A. Schematic of fibrinogen showing fibrinopeptides A (FpA) and B (FpB), the constitutive holes “a” and “b” in the globular C-terminal domains of the γ chains and β chains, respectively, and the “A” and “B” knobs, which are exposed only after FpA and FpB cleavage by thrombin. Here the globular βC and γC domains are shown separately, βC in purple, γC in blue as in Fig. 125–2. B. Self-assembly of fibrin units to form an organized polymeric structure. Here, for simplicity, the D regions are represented as a single globular unit.
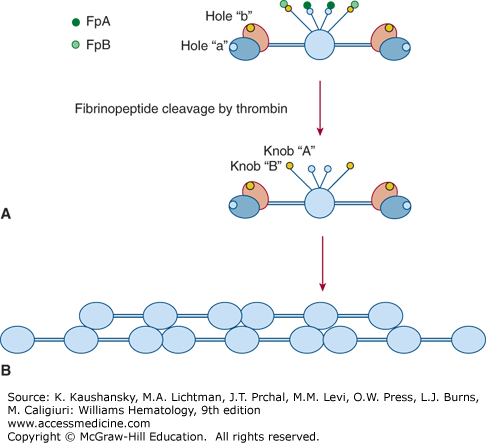
A knob-hole a (A:a) interaction results in formation of double-stranded fibrils in which fibrin molecules become aligned in an end-to-middle, staggered overlapping arrangement (see Fig. 125–3).16,17,18 Fibrils subsequently undergo branching by lateral fibril associations in which two fibrils converge to form a four-stranded “bilateral” fibril junction. Progressive lateral associations among fibrils result in larger fibril bundles or fibers. A second type of junction, termed equilateral branching, is formed by three fibrils converging to form a three-member junction.19 Both types of branch junctions provide scaffolding for the clot network, the ultimate structure of which is governed by several variables, including salt concentration, pH, fibrinogen concentration and thrombin concentration.16,17,20
Fibrinopeptide B (FpB) release occurs more slowly than fibrinopeptide A (FpA) release and exposes another polymerization site known as the “B” knob beginning with the amino acid sequence GHRP. GHRP interacts with a constitutive hole “b” in the β chain encompassed by residues 427 to 462 (397 to 432). FpB cleavage is accelerated by fibrin polymerization, whereas FpA cleavage is independent of fibrin polymerization. B:b interactions are not required for lateral fibril associations, but they contribute to lateral association by inducing rearrangements in βC that allow βC:βC contacts to occur.21,22
The flexible αC domains also participate in fibrin polymerization.23 Fibrin clots made from plasma fibrinogen molecules lacking more than 100 C-terminal residues from the αC domain display prolonged thrombin times, reduced turbidity, and produce thinner fibers, indicating that αC domains participate in lateral fibril associations. In addition, αC domains become dissociated as a result of FpB cleavage. This allows αC domains to participate in noncovalent interactions with other αC domains, thereby promoting lateral fibril associations and fibrin network assembly. Finally, additional self-associating sites in the D region participate in fibrin assembly. These are the D:D sites and γXL sites that promote end-to-end alignment of assembling fibrin units and factor XIIIa crosslinking, respectively.24,25
*The recommendation of the Human Genome Variation Society (HGVS) is to number amino acid residues from the initiator Met, with the protein reference sequences representing the primary translation product, not the processed, mature, protein. This is the standard nomenclature used by geneticists. For fibrinogen, however, as for many other secreted proteins, such as the coagulation factors, this is not the nomenclature used in earlier publications (historically fibrinogen residues are numbered according to the secreted product lacking the signal peptide). In this text both nomenclatures are used: amino acid residues and substitutions are described first according to HGVS guidelines followed in brackets by the corresponding amino acid in the mature chain lacking the signal peptide. To convert from the HGVS nomenclature to the mature protein nomenclature, subtract 19 for Aα, 30 for Bβ, or 26 for γ. A one-letter abbreviation for amino acids is used in this chapter. A, alanine; C, cysteine; D, aspartic acid; E, glutamic acid; F, phenylalanine; G, glycine; H, histidine; I, isoleucine; K, lysine; L, leucine; M, methionine; N, asparagine; P, proline; Q, glutamine; R, arginine; S, serine; T, threonine; V, valine; W, tryptophan; Y, tyrosine.
CROSSLINKING BY FACTOR XIII
The clot formed by fibrin polymerization requires further stabilization to increase its mechanical strength and resist immediate degradation by the fibrinolytic pathway. Factor XIIIa (a heterotetramer FXIII-A2B2) is a transglutaminase that stabilizes the elongating protofibril by crosslinking adjacent γ chains through the formation of ε-(γ-glutamyl) lysine isopeptide bonds.26 These occur between lysine 432 (406) of one γ chain and glutamine 424 (398) or 425 (399) of another chain. Crosslinking increases the resistance of the clot to deformation. The same process occurs, but at lower rate, between α chains and also between α chains and γ chains. In the presence of factor XIIIa, α-antiplasmin becomes covalently bound to the distal α chains of fibrin or fibrinogen.26 The factor XIII binding site for fibrin has been characterized: residues in the Aα-C domain, that is, 408 to 421 (389 to 402) bind a cleft in FXIII-A2 that is exposed only after cleavage of the activation peptide by thrombin.27 Fibronectin is also incorporated into the fibrin clot. This occurs by noncovalent interactions between the two proteins through specific binding sites, followed by their covalent crosslinking with factor XIIIa.28 Fibronectin incorporation appears to affect the adhesion and migration of cells at sites of fibrin deposition, thereby contributing to wound healing and other cell-dependent processes.
FIBRINOLYSIS
Plasminogen and tissue-type plasminogen activator (t-PA) binding sites in the D regions (i.e., γ 337 to 350) (312 to 324), and αC domains (i.e., Aα 167 to 179) (148 to 160), are cryptic in fibrinogen and become exposed during fibrin assembly or during formation of crosslinked fibrinogen fibrils (Chap. 135).29,30 Two phases can be distinguished in the t-PA induced lysis of a fibrin clot.31. In the first, slow, phase, t-PA activates plasminogen on the intact fibrin surface. The generation of C-terminal lysine residues in partially degraded fibrin (by plasmin) in the second phase of clot lysis may result in accumulation of plasminogen at the clot surface and a concomitant increase in lysis rate. Thrombin-activatable fibrinolysis inhibitor (TAFI) removes C-terminal lysine residues, resulting in a strongly reduced binding of plasminogen and in an inhibition of the second phase of clot lysis by a reduction of the activation of plasminogen on the fibrin surface. TAFI, as well as α-antiplasmin, lipoprotein(a), and histidine-rich glycoprotein, bind to fibrin and all have an inhibitory effect on fibrinolysis through various mechanisms.
ANTITHROMBIN ACTIVITY OF FIBRIN
Thrombin binds to its substrate, fibrinogen, through a fibrinogen recognition site in thrombin, referred to as exosite 1. The fibrin clot itself also exhibits significant thrombin-binding potential; this nonsubstrate binding potential of fibrin for thrombin is referred to as antithrombin activity I.6 This activity is defined by two classes of nonsubstrate thrombin-binding sites in fibrin, one of “low-affinity” in the E-region and the other of “high-affinity” in D regions of fibrin(ogen) molecules containing the variant γ′ chain. Altogether, heterodimeric γA/γ′ and homodimeric molecules γ′/γ′ chains make up 8 to 15 percent of the total γ-chain population.10 Low-affinity thrombin-binding activity reflects thrombin exosite 1 binding in the E region of fibrin, whereas high-affinity thrombin binding to γ′ chains takes place through exosite 2. The binding affinity of thrombin for γ′-containing fibrin molecules is increased by concomitant fibrin binding to thrombin exosite 1. Antithrombin I (fibrin) is an important inhibitor of thrombin generation that functions by sequestering thrombin in the forming fibrin clot, and also by reducing the catalytic activity of fibrin-bound thrombin. Vascular thrombosis may result from absence of antithrombin I (as in afibrinogenemia; see “Afibrinogenemia and Hypofibrinogenemia” below), reduced plasma γ′-chain content,32 or defective thrombin binding to fibrin as found in certain dysfibrinogenemias (see “Dysfibrinogenemia and Hypodysfibrinogenemia” below). In contrast, an increased susceptibility to arterial thrombosis has been reported when γ′-chain levels are significantly elevated. Moreover, thrombin bound to γA/γ′-fibrin is protected from inhibition by antithrombin to a greater extent than thrombin bound to γA/γA-fibrin. Thus, γA/γ′-fibrin serves as a reservoir of active thrombin, which may contribute to the prothrombotic nature of thrombi.33
AFIBRINOGENEMIA AND HYPOFIBRINOGENEMIA
Type I disorders (afibrinogenemia and hypofibrinogenemia) affect the quantity of fibrinogen in circulation. Type II disorders (dysfibrinogenemia and hypodysfibrinogenemia) affect the quality of circulating fibrinogen.1 While the first dysfibrinogenemia mutation was identified as early as 1968,34 the molecular basis of afibrinogenemia was elucidated much later.35 This disorder is characterized by autosomal recessive inheritance and the complete absence of fibrinogen in plasma.
The disease, originally described in 1920,36 has an estimated prevalence of approximately one in 1,000,000. In populations where consanguineous marriages are common, the prevalence of afibrinogenemia, is increased.37 Because hypofibrinogenemia (fibrinogen levels below 1.5 g L−1) is often caused by heterozygosity for a fibrinogen gene mutation, this is much more frequent than afibrinogenemia. If one applies the Hardy Weinberg binomial distribution of alleles in the population to afibrinogenemia, carriers of fibrinogen deficiency causing mutations could be as frequent as 1 in 500.
Since the identification of the first causative mutation for congenital afibrinogenemia in 1999,35 approximately 100 distinct mutations, the majority in FGA, have been identified in patients with afibrinogenemia (in homozygosity or in compound heterozygosity) or in hypofibrinogenemia. Causative mutations can be divided into two main classes: null mutations with no protein production at all and mutations producing abnormal protein chains which are retained inside the cell.1
The first causative mutation for afibrinogenemia was identified in a nonconsanguineous Swiss family with two pairs of afibrinogenemic brothers.35 In a first step toward establishing whether or not the disease was linked to the fibrinogen gene cluster on chromosome 4, haplotype data were obtained for five microsatellite markers surrounding this locus. One of these, FGAi3, a (TCTT)n polymorphic marker located in intron 3 of the FGA gene, was found to be deleted in all four affected individuals and was hemizygous in the obligate carriers, implying that homozygous deletion of at least part of the FGA gene was responsible for the congenital afibrinogenemia in this family. Indeed, the genetic defect was found to be a recurrent deletion of approximately 11 kb of DNA, with breakpoints in FGA intron 1 and the FGA–FGB intergenic region, resulting in an absence of fibrinogen.
Three other large deletions in the fibrinogen gene cluster have been identified, all involving part of the FGA gene. These are: a deletion of 1.2 kb eliminating the entire FGA exon 4 in a Japanese patient38; a deletion of 15 kb, with breakpoints situated in FGA intron 4 and in the FGA–FGB intergenic region in a Thai patient39; and a 4.1-kb deletion encompassing FGA exon 1 in an Italian patient.40 All patients were homozygous for the identified deletions except for the Thai patient, for whom complete maternal uniparental disomy was confirmed for the deleted chromosome 4.39
Several splice-site mutations have been identified in all three fibrinogen genes. In afibrinogenemic patients of European origin, the most common mutation is a donor splice mutation in intron 4, c.510+1G→T (previously described as IVS4+1 G→T).1,41 Haplotype data suggest that this mutation, like the FGA 11-kb deletion, is also recurrent, or a very ancient mutation, because the c.510+1G→T mutation is found on multiple discrete haplotypes.
Frameshift mutations have been identified in all three fibrinogen genes. FGA exon 5, the largest fibrinogen-coding exon has the most frameshift mutations. Interestingly, seven single base-pair deletions in FGA
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


