Variant class
Pathogenicity
1
Pathogenic
2
Likely pathogenic
3
Uncertain significance
4
Likely benign
5
Benign
Mutations may be described as either constitutional or somatic. Constitutional mutations are also known as germline mutations. These are present in gametes prior to fertilisation and therefore present in every cell of the individual resulting from that embryo. They are highly heritable between generations. Germline mutations occurring in an oncogene or tumour suppressor gene (TSG) cause an inherited cancer predisposition and the individual will usually have a high lifetime risk of cancer development.
Most cancers, however, develop from somatic mutations, which arise in dividing cells post-fertilisation. Somatic mutations are present only in descendants of that cell and are not heritable between generations [10].
5.3 Somatic Mutations in Oncogenesis
Individuals accrue somatic mutations in cells throughout their lifetime (◘ Fig. 5.1). In a tumour cell, somatic mutations are identified as either driver or passenger mutations. A driver mutation is a cancer-causing mutation, which gives the cell a selective growth advantage in tumourigenesis. Passenger mutations co-exist alongside driver mutations but do not contribute to cancer development and confer no survival benefit. Both driver and passenger mutations are replicated throughout the clonal population descending from the initial tumour cell. Chemotherapy resistance mutations begin as passenger mutations, initially providing no advantage to the growing tumour. Once challenged with chemotherapy, they offer a selective advantage upon which a subclone can develop and can then be considered driver mutations. This may manifest phenotypically as cancer recurrence [10].
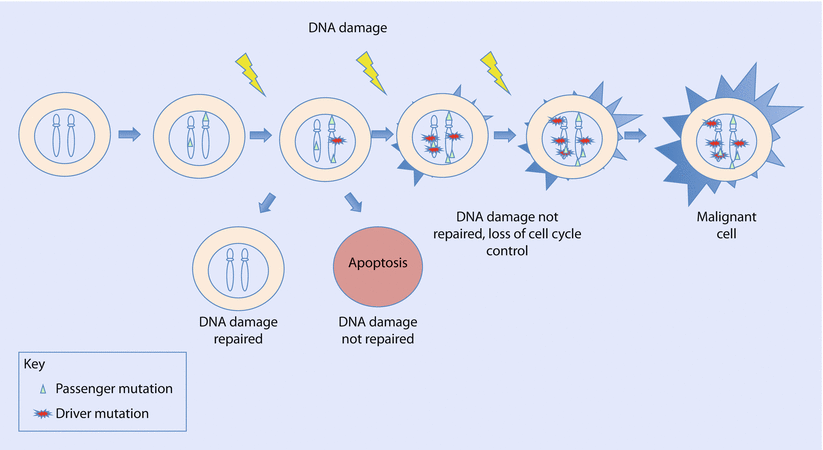
Fig. 5.1
The cell accumulates both passenger and driver somatic mutations which are replicated in a clonal population with each mitosis. Driver mutations contribute to cancer development. The cell may repair DNA damage or trigger cell death by apoptosis. If DNA damage repair is deficient, mutations accumulate causing genomic instability and loss of cell cycle control, and the cell acquires malignant potential
Intratumoural heterogeneity is a feature of breast tumour tissues, in which several genetically diverse subclones of cells exist within the tumour tissue. For example, The Cancer Genome Atlas performed whole exome sequencing of 510 breast tumours and identified over 30,000 somatic mutations [11]. Of these, they found that mutations in only three genes, PIK3CA, TP53 and MAP3K1, occurred in more than 10% tumours. Cells with mutations offering the greatest advantage to the growing tumour are selected by way of evolutionary pressure [12]. Gene expression in breast cancer tumours may also be modified by epigenetic changes, which lead to aberrant gene regulation without altering the DNA sequence. Examples of epigenetic changes include hypermethylation of gene promotor regions or changes to the structure of the DNA-protein packaging complex, chromatin. Both of these mechanisms may lead to silencing of tumour suppressor genes (TSGs) [12].
Somatic mutations in cancer are collated in the Catalogue Of Somatic Mutations In Cancer (COSMIC) database (► http://cancer.sanger.ac.uk/cosmic) [13]. This is a publicly searchable online database, which combines mutation data with evidence from the published scientific literature [14].
Somatic mutations in breast cancer may be useful indicators of treatment response and prognosis. However, intratumoural heterogeneity poses a challenge for tumour profiling and may explain why some patients fail to respond to targeted treatments. For example, the American Society of Clinical Oncology (ASCO) and the College of American Pathologists recommend that molecular profiling of breast tumours for oestrogen and progesterone receptor positivity requires only 1% of tumour nuclei to express positivity by immunohistochemistry [15] which, it may be argued, fails to take into account the variation within the tumour. Furthermore, discrepancies exist between the molecular profiles of primary and metastatic tumours suggesting a need for multiple biopsies during systemic treatment to optimise treatment design and minimise the emergence of chemotherapy-resistant clones [12].
5.4 Constitutional Mutations in Oncogenesis
Hereditary cancers occur due to a constitutional mutation in a gene critical to cell cycle regulation with a second driver mutation triggering tumourigenesis.
Alfred Knudson first provided evidence in 1971 for hereditary cancer syndromes by studying cases of familial retinoblastoma [16, 17]. He found that individuals with bilateral retinoblastomas developed these at a younger age than individuals with unilateral retinoblastomas. Not all individuals with a family history developed retinoblastomas, yet retinoblastomas could be observed in their offspring. This was suggestive of the presence of a germline mutation for which a further «hit» or mutation was required for tumour development. Where unilateral cases occurred at a later age, these were due to two somatic mutations, which took more time to accumulate. Knudson proposed his «two-hit hypothesis» in which two loss-of-function mutations were required for tumour development (◘ Fig. 5.2). RB1 was later identified as the first tumour suppressor gene (TSG).
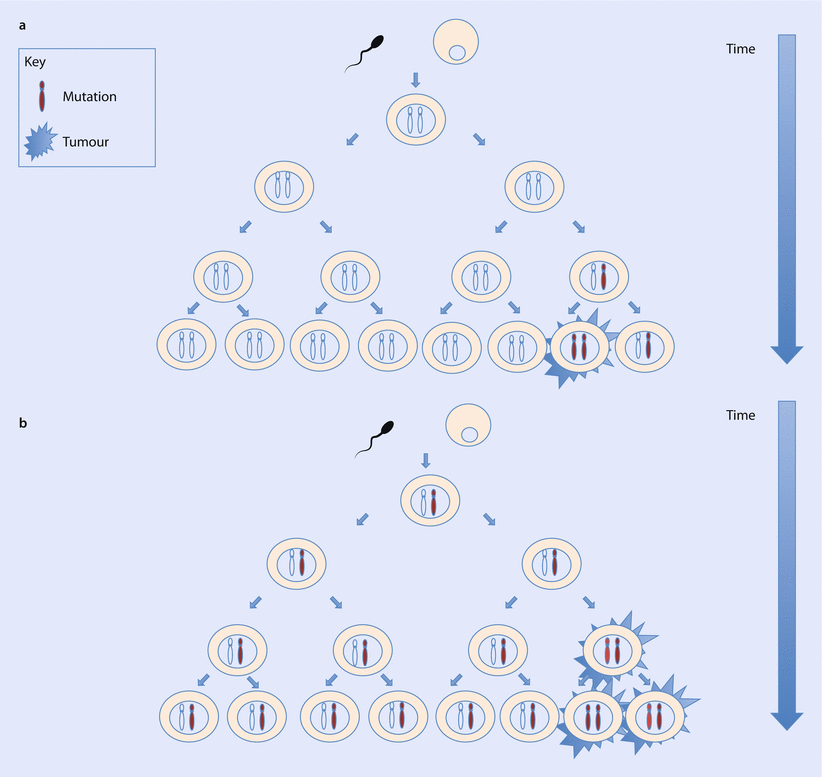
Fig. 5.2
a Non-hereditary cancer. Somatic mutations occur in cells post-fertilisation, with a later age at cancer diagnosis. b Hereditary cancer. Germline mutation present in all cells from conception. Only one somatic mutation is required for tumour initiation, resulting in earlier onset of disease
Although it is now believed that multiple hits may be required for cancer development, Knudson’s model still forms the basis for understanding the mechanism of cancer development in inherited cancer syndromes.
5.5 Hereditary Breast Cancer Susceptibility
The Utah Population Database demonstrated an increased familial risk of breast cancer in first-, second- and third-degree relatives compared to population risk, providing evidence for a significant genetic contribution to breast cancer development [18]. Genes implicated in the development of breast cancer can be considered in terms of their penetrance, or their likelihood of causing disease, as high, moderate or low penetrance. High-penetrance genes are rare and typically cause a greater than fourfold increased risk compared to the general population (◘ Table 5.2). Moderate-penetrance genes cause a two-to fourfold increased risk with the remainder being more common low-penetrance alleles (◘ Fig. 5.3) [19]. This corresponds with the UK National Institute of Health and Care Excellence’s (NICE) definitions, where those at high risk have a lifetime breast cancer risk of >30% and those at moderate risk have a lifetime breast cancer risk of 17–29% [20].

Table 5.2
Summary of clinical features of high-penetrance hereditary breast cancer genes
Gene | Syndrome | Population frequency | Breast cancer risk | Median age | Special tumour type | Other features | |
|---|---|---|---|---|---|---|---|
BRCA1 and BRCA2 | Hereditary breast and ovarian cancer | 1/400 to 1/800 | High | BRCA1 60% by age 70 (95% CI 44–75%) BRCA2 55% by age 70 (95% CI 41–70%) | BRCA1 42 years BRCA2 45 years | BRCA1 triple-negative, basal tumours | Ovarian cancer Pancreatic cancer Prostate cancer (males) |
TP53 | Li-Fraumeni syndrome | 1/5000 to 1/20,000 | High | a | 33 years | Often HER2 positive | Sarcoma Brain tumours Adrenocortical carcinomas May have multiple primaries |
STK11 | Peutz-Jeghers syndrome | 1/155,000 | High | 45% by age 70 (95% CI 27–68%) | 44 years | Mucocutaneous pigmentation Hamartomatous polyps Increased risk of GI, ovarian, pancreatic cancer and adenoma malignum of uterine cervix Gonadal tumours | |
PTEN | Cowden syndrome | 1/200,000 | High | 77% by age 70 (95% CI 59–91%) | 42 years | Increased incidence of benign breast disease | Macrocephaly Learning difficulties Thyroid cancer Endometrial cancer Gastrointestinal cancer Renal cell cancer |
CDH1 | Hereditary diffuse gastric cancer | Unknown | High | 39% by age 80 (95% CI 12–84%) | 53 years | Invasive lobular breast cancer | Gastric cancer |
Fig. 5.3
Schematic representation of genetic modifiers of breast cancer risk. Genes can be grouped into rare, high-penetrance mutations, moderate-penetrance mutations and low-penetrance alleles. The approximate location of known genes is shown with respect to allele frequency and relative risk
The majority of genes implicated in hereditary breast cancers occur due to protein truncating mutations in genes active in the DNA damage repair pathway [19]. Breast cancer susceptibility genes have been identified using three main techniques: linkage analysis studies, mutation screening of candidate genes and genome-wide association studies (GWAS). Linkage analysis studies are useful in large families with multiple affected relatives whereby regions of the genome co-segregating in affected family members can be analysed for highly penetrant cancer susceptibility genes [21]. Mutation screening of candidate genes has developed alongside our understanding of oncogenesis and, in particular, the DNA damage repair pathway.
5.6 High-Penetrance Genes
5.6.1 BRCA1 and BRCA2
The BRCA1 and BRCA2 genes were identified as breast cancer predisposition genes in the mid-1990s by linkage analysis studies [22, 23]. In an intact DNA damage repair response, the BRCA1 and BRCA2 proteins form part of a complex recruited to the site of DNA double strand breaks to accurately repair DNA damage by homologous recombination. Homologous recombination uses an intact complementary strand as a template to faithfully restore the DNA sequence. In contrast, BRCA-deficient cells repair DNA damage by non-homologous repair pathways, prone to error, causing genomic instability [24, 25].
BRCA1 and BRCA2 carrier frequency is estimated to be around 1/400 to 1/800 in the general population [26–28]. In certain populations, carrier frequency may be significantly higher due to a founder effect, whereby rare mutations are seen at relatively high frequencies within small, genetically isolated populations. One example is the Ashkenazi Jewish population, in which three common founder mutations, BRCA1_185delAG, BRCA1_5382insC and BRCA2_6174delT, combine to give an overall carrier risk of 1 in 40 in the Ashkenazi Jewish population. [29, 30]. The proportion of hereditary breast cancers in this group is significantly elevated compared to that of the general population. In Iceland, the BRCA2_999del5 founder mutation occurs in 0.4% of the population and is responsible for the majority of inherited breast cancer risk in this country, accounting for 8.5% breast cancers, 7.9% ovarian cancers and 2.7% prostate cancers [31]. It should be noted that founder mutations in populations from resource-poor settings are less well described.
A prospective analysis as part of the EMBRACE study reported breast cancer risk by age 70 as 60% (95% confidence interval (CI) 44–75%) in BRCA1 and 55% (95% CI 41–70%) in BRCA2 carriers, respectively. Ovarian cancer risk was 59% (95% CI 43–76%) in BRCA1 and 16.5% (95% CI 7.5–34%) in BRCA2 carriers [32]. Median age at diagnosis of breast cancer in BRCA1 mutation carriers was 42 years and in BRCA2 mutation carriers, 45 years [32].
Within the BRCA1 and BRCA2 genes, thousands of variants have been identified, occurring across the entire coding regions of the gene. Most pathogenic mutations involve the formation of a truncated BRCA1 or BRCA2 protein and occur as a result of non-sense mutations, frameshift mutations, splice site mutations or larger-scale rearrangements [21, 33].
Pathologically, invasive breast tumours in BRCA1 mutation carriers often display a basal epithelial phenotype [34] and characteristically stain negative by immunohistochemistry for oestrogen receptors (ER), progesterone receptors (PR) and human epidermal growth factor receptor 2 (HER2), so-called triple-negative tumours (TNT). A large analysis of breast tumour datasets showed that ER positivity was negatively predictive of BRCA1 mutations. TNTs were identified in 67.3% of BRCA1 mutation carriers under 50 years old and 57.7% in those over 50 years and in 13% of BRCA2 mutation carriers under 50% and 23.5% in those over 50 years. TNTs were highly predictive of BRCA1 mutations at any age and modestly predictive of BRCA2 mutations in individuals over 50 years [35].
5.6.2 TP53
Germline loss-of-function mutations in the tumour suppressor gene, TP53, cause the cancer predisposition syndrome Li-Fraumeni Syndrome (LFS) [36]. LFS is characterised by childhood- and adult-onset tumours comprising soft tissue and bone sarcomas, early-onset breast cancer, brain (especially choroid plexus) tumours and adrenocortical carcinomas, although the spectrum of reported tumours is diverse. One report calculated a population frequency of TP53 germline mutations as 1 in 5000 [37] although more conservative estimates have suggested this is closer to 1 in 20,000 [38], with a de novo mutation rate of between 7% and 20% [39]. Lifetime cancer risk in LFS approaches 100% in females and 73% in males. This gender discrepancy is largely due to the high risk of female breast cancer in LFS [40]. Breast cancer in LFS typically occurs in premenopausal females, with an average age of onset of 33 years [41]. The risk ratio for breast cancer development in TP53 mutation carriers is 6.4 (95% CI 4.3–9.3) [42], and 31% of carriers develop a further tumour in the contralateral breast [41]. Pathologically, breast tumours in TP53 germline mutation carriers tend to demonstrate HER2 overexpression [43]. TP53 mutation carriers may develop multiple primaries, with approximately 30% developing a second tumour within a radiation field within a mean of 10.7 years, suggesting the need for carefully scheduled treatments plans in LFS [41].
The TP53 mutation carrier rate in women with early-onset breast cancer (<30 years of age), who have no family history of relevant cancers, is estimated to be between 5% and 8%, and McCuaig and colleagues (2012) recommend BRCA1/2-negative breast cancer patients under 30 years be offered TP53 testing [44]. The Chompret Criteria can also be used to indicate individuals appropriate for TP53 mutation testing. Recent revisions to the Chompret Criteria recommend testing is based on family history, a personal history of breast cancer under age 31, the occurrence of multiple primary tumours and the occurrence of rare tumours [41].
5.6.3 STK11
Germline mutations in the tumour suppressor gene, STK11 (previously known as LKB1), give rise to Peutz-Jeghers syndrome (PJS). STK11 is a serine threonine kinase involved in cell polarity and regulation of downstream effectors of the mTOR (mammalian target of rapamycin) signalling pathway [45]. Population frequency is believed to be around 1 in 155,000 although estimates vary [46]. Peutz-Jeghers syndrome (PJS) is characteristically associated with mucocutaneous pigmentation, hamartomatous polyps and an increased risk of a variety of cancers, which may include gastrointestinal, pancreatic, breast, gonadal and gynaecological cancers. Female breast cancer risk in PJS is 45% (95% CI 27–68%) by age 70 years with an overall cancer risk of 85% (95% CI 68–96%) [47]. A meta-analysis suggested an average age of onset of breast cancer in STK11 carriers of 44 years [48].
5.6.4 PTEN
PTEN is a tumour suppressor gene, which antagonises and suppresses the PI3K-AKT-mTOR pathway [49]. Germline mutations in PTEN cause PTEN hamartoma tumour syndrome, which includes Cowden syndrome (CS) [50]. Prevalence is believed to be between 1 in 200,000 and 1 in 250,000 [51]. CS is an autosomal dominant hamartoma syndrome comprising macrocephaly, mucocutaneous lesions and an increased benign and malignant tumour risk, particularly thyroid, breast, endometrial, renal and colorectal cancers [52]. Benign breast disease is also found in PTEN gene mutation carriers. A cohort of 154 PTEN mutation carriers found that 47% of the 55 female carriers had fibrocystic breast disease. Breast cancer occurred in 34% of female carriers, occurring at a median age of 42 years, with 48% of breast cancers occurring bilaterally. The cumulative risk of breast cancer by age 70 was 77% (95% CI 59–91%) [53]. In an international, multicentre cohort of germline PTEN mutation carriers, the cumulative risk of breast cancer was 67% by age 60 years, giving a five- to sevenfold increased risk compared to the general population [54].
5.6.5 CDH1
CDH1 encodes the glycoprotein E-cadherin, which has an important role in maintaining cell-cell adhesion and cell polarity [55]. Germline CDH1 mutations were identified by linkage analysis to cause hereditary diffuse gastric cancer [56]. Germline CDH1 mutations have also been associated with development of lobular breast cancer, with a cumulative breast cancer risk by age 80 of 39% (95% CI 12–84%), giving a risk ratio of 6.6 [57]. This older data is supported by a 2015 study of breast cancer risk in individuals with CDH1 germline mutations which estimated breast cancer risk of 42% by age 80 (95% CI 23–68%) [58]. A recent review by Corso and colleagues (2016) identified 14 reported cases of CDH1-associated breast cancer without a family history of diffuse gastric cancer and suggested the concept of «hereditary lobular breast cancer» as its own clinical entity [55].
5.7 Moderate-Penetrance Mutations
Several genes have been proposed as harbouring moderate-penetrance mutations.
5.7.1 PALB2
PALB2 interacts with BRCA1 and BRCA2 at the site of DNA damage as part of DNA damage repair by homologous recombination [59]. Bi-allelic mutations in PALB2 are associated with the childhood autosomal recessive condition, Fanconi anaemia, whilst germline heterozygous mutations in PALB2 are associated with an increased risk of breast cancer. Risk of breast cancer in PALB2 mutation carriers borders the moderate- to high-penetrance risk group. Antoniou and colleagues (2014) suggested PALB2 mutations confer a high risk of breast cancer, finding an up to ninefold increased risk in mutation carriers under 40 and a cumulative lifetime risk of 35% (95% CI 26–46%) by age 70 [60]. However, age, birth cohort and family history influenced this risk. Their analysis suggested breast cancer risk was modified by other genetic and environmental factors precluding attempts to provide a single estimate of breast cancer likelihood in all mutation carriers and leading to the suggestion that family history and genotype be considered in risk estimations. Other studies have estimated a lower risk, and wide confidence intervals noted on meta-analysis mean that PALB2 is still considered a moderate-penetrance allele [19].
5.7.2 ATM
The ataxia telangiectasia mutated (ATM) gene is activated in response to DNA damage. ATM regulates downstream targets, including, amongst others, p53 and CHEK2, important in cell cycle regulation, and BRCA1-mediated DNA damage repair [61]. Bi-allelic loss-of-function mutations in ATM cause the autosomal recessive condition, ataxia telangiectasia, characterised by ataxia, immune dysfunction, radiation sensitivity and cancer predisposition. Heterozygote loss-of-function mutations in ATM have been associated with increased risk of breast cancer. A recent meta-analysis of 19 studies by Marabelli and colleagues (2016) estimated the cumulative breast cancer risk in heterozygote ATM carriers to be 32.8% by age 80 (95% CI 24.5–40.3%) [62].
5.7.3 CHEK2
Constitutional mutations in CHEK2 are associated with increased breast cancer risk. A protein truncating founder mutation, CHEK2_1100delC, has been identified in North European populations and is estimated to confer a twofold increased breast cancer risk in women [63]. A recent report has suggested it may also increase the risk of gastric, renal, sarcoma and prostate cancer [64].
Other genes, such as MRE11, NBN and RAD50, exist within the same DNA damage repair system and have been implicated as increasing breast cancer risk although this evidence of association is more limited.
5.8 Low-Penetrance Alleles
Low-penetrance variants account for around 14% of the genetic contribution to breast cancer [65]. These variants occur with relatively common frequency within the general population and are expected to operate in polygenic models with multiple weak associations contributing to cancer risk [66]. Pathogenic variants have been identified in CASP8 and TFGR1 genes. In addition, multiple susceptibility loci have been identified, some of which can be mapped to individual genes, such as FGFR3 and TNRC9. However, other loci map to several genes and some loci cannot be mapped to gene coding regions at all. The mechanisms by which these susceptibility loci function in breast cancer risk are incompletely understood [21].
5.9 Risk Prediction
All individuals with breast cancer and particularly those at risk of a hereditary cancer predisposition syndrome should have their risk assessed through a three-generation family history [67]. This may be conducted as part of assessment at a specialist breast cancer family history clinic or clinical genetics consultation. Referral guidelines to clinical genetics services for suspected hereditary breast cancer are available in the UK from NICE [20] and in the USA, from the National Comprehensive Cancer Network (NCCN) [68]. The consensus from the European 14th St Gallen International Breast Cancer Conference is that individuals with breast cancer diagnosed at less than 40 years, those with a strong family history, or individuals diagnosed at less than 50 years with a triple-negative tumour, without a family history of breast cancer, should be considered for BRCA1 and BRCA2 testing [69].
Decision support tools have been developed to enable clinicians to identify patients who need further assessment by clinical genetics services. For example, Guy’s and St Thomas’ regional genetics service has developed an interactive decision support tool and hereditary cancer reference guide, accessible as a smartphone application («App») or via desktop (► www.ubqo.com/cancergenetics) [70]. This aims to simplify the referral process so that at risk individuals can be identified and appropriately referred whilst individuals with low risk can be reassured.
5.10 Tools in Risk Prediction
The Manchester scoring system (MSS) uses information from the family history to calculate the probability of finding a pathogenic BRCA1 or BRCA2 mutation [71]. An updated model was proposed in 2009, which incorporates tumour histology and hormone receptor status [72]. The MSS has the advantage of being quick and simple to use in a clinical setting but is not recommended for use in founder populations or populations in which breast cancer is rare [71]. The UK NICE guidelines recommend BRCA1 and BRCA2 testing in individuals with a carrier probability of ≥10%, which equates to a MSS score of ≥15 [20].
In addition, various computerised models have been designed. Examples of such models include Claus, Gail, Tyrer-Cuzick or IBIS, BRCAPro and BOADICEA. All models are limited by accessibility, the requirement for user data entry, which can be time-consuming and the reliance on self-reported family history information, which may be limited or inaccurate [73]. The models are calibrated to different population datasets and incorporate additional risk factor assessments to different degrees.
The Gail model was proposed in 1989, based on data from the Breast Cancer Detection Demonstration Project (BCDDP) in which data was gathered for approximately 300,000 individuals undergoing annual mammographic breast cancer screening [74]. This model incorporates six risk factors, including age, age at menarche and first live birth, number of previous breast biopsies, history of atypical hyperplasia and breast cancer occurrence in first-degree relatives. The original model predicts the probability of in situ and invasive breast cancer development in an individual undergoing annual screening, but updates to this model predict the absolute risk of invasive breast cancer occurrence and in addition adjust the model to predict cancer occurrence in black women [75]. Neither model accounts for high-risk genes such as BRCA1 or BRCA2 mutation carriers. A systematic review demonstrated that the early Gail models had limited ability to predict individualised risk [76], possibly due to its failure to account for high-risk groups [73]. A systematic review of the Gail model with further updates has demonstrated improved accuracy compared to the original model [77].
The Claus Model, proposed in 1991, uses data from the Cancer and Steroid Hormone study, a case-control study of 4730 confirmed breast cases [78]. The model combines information on breast cancer occurrence in mothers and sisters of index cases and the age at diagnosis. This is used to estimate which cases are expected to be high-risk gene mutation carriers and the estimated age-specific and cumulative lifetime risk of breast cancer development in carriers. The model was updated in 1993 to include the occurrence of ovarian cancer in the family history [79]. The dataset is restricted to white, North American subjects, which may limit its widespread generalisability, and it does not address the extended family history or other recognised breast cancer risk factors [73].
Following the identification of BRCA1 and BRCA2, the Tyrer-Cuzick or IBIS model was developed to incorporate genetic risk factors with hormonal and reproductive risk factors to predict individual breast cancer risk. Genetic risk was based upon the possibility of a BRCA1 or BRCA2 mutation plus a second low-penetrance gene to acknowledge and account for other unknown influencers of genetic risk. Family history included first- and second-degree relatives with a history of breast or ovarian cancer, bilateral breast cancer and the age at diagnosis. Additional personal risk factors included age, height, body mass index (BMI), age at menarche, first live birth and menopause, parity and the presence of atypical hyperplasia or lobular carcinoma in situ [80]. It has recently (2013) been updated to include the impact of Ashkenazi Jewish ancestry, third-degree relatives, male breast cancers and hormone replacement therapy use. It calculates the lifetime risk of breast cancer and the risk of gene carriage.
The BRCAPro model was developed to determine the likelihood of being a BRCA1 or BRCA2 mutation carrier [81]. This model uses detailed information in the family history such as breast cancer in first- and second-degree relatives, ages at diagnoses, bilateral breast cancer, ovarian cancer and the presence of male breast cancers. In addition, the model also gives weight to unaffected relatives. Extensions to the model enable an overall risk of breast cancer development to be calculated, and three different population datasets can be used to estimate mutation frequency [73]. Limitations to the model include the absence of non-hereditary risk factors, and the model does not account for more complex genetic susceptibility mechanisms.
The Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm (BOADICEA) model was developed to account for genetic and polygenic influences in familial breast cancer occurring in addition to BRCA1 and BRCA2 [82, 83]. The dataset was based on the Anglican Breast Cancer Study, which comprised 1484 confirmed breast cancer cases and 156 multiple case families and included information about BRCA1 and BRCA2 mutation status. The model included detailed family history information including breast cancer diagnoses in first-, second- and third-degree relatives and ages at diagnoses, bilateral cancer, ovarian cancer and male breast cancer. Aside from age, it did not include any non-hereditary risk factors. BOADICEA provides likelihood of breast cancer development in women with a family history of breast and ovarian cancer. In addition, the model can predict BRCA1 and BRCA2 mutation probability and provides an assessment of polygenic modifiers of breast cancer risk in mutation carriers. An extension to the BOADICEA model has recently been proposed to incorporate PALB2, CHEK2 and ATM protein truncating variants into its risk assessment [84].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


