INTRODUCTION
SUMMARY
Heparin-induced thrombocytopenia (HIT) is a prothrombotic complication of treatment with heparin. It is associated with mild-to-moderate thrombocytopenia, although the main clinical concern is the high frequency of both arterial and venous thromboembolism, which may be limb- or life-threatening. HIT is an immune complex-based disorder involving platelet factor 4 complexed to negatively charged multimeric molecules, especially surface heparan side chains. It is initiated by exposure to heparin, particularly unfractionated heparin. There is growing understanding of the unusual nature of the underlying immune response in HIT, why certain individuals develop this disorder, and why HIT is prothrombotic. Diagnosis is based upon an assessment of clinical probability and specialized laboratory testing. Management involves immediate cessation of heparin and initiation of parenteral inhibitors of thrombin or factor Xa.
Acronyms and Abbreviations
CI, confidence interval; DIC, disseminated intravascular coagulation; GAG, glycosaminoglycan; HIT, heparin-induced thrombocytopenia; INR, international normalized ratio; LMWH, low-molecular-weight heparin; MTHFR, methylenetetrahydrofolate reductase; PF4, platelet factor 4; UFH, unfractionated heparin.
DEFINITION AND HISTORY
Heparin-induced thrombocytopenia (HIT) is a complication of heparin therapy in which there is a fall in platelet count and an unusually high incidence of arterial and/or venous thromboembolic complications in association with heparin therapy.
Although clinical usage of heparin as an anticoagulant began in the late 1950s, it was not until the early 1970s that a small percentage of treated patients were noted to develop a complication consisting of thrombocytopenia with paradoxical, life-threatening thromboemboli (for a historical review see Ref. 1). In the 1980s, it became clear that HIT was caused by immunoglobulin (Ig) G antibodies that activate platelets. It was also recognized that HIT could be divided into two types, the classic immune-mediated prothrombotic disease that is the focus of this chapter (formerly called HIT type II), and a benign nonimmune condition associated with a mild, immediate, and transient drop in platelet count and no increased risk of thrombosis (formerly called HIT type I).2 In this chapter, “HIT” means the immune-mediated form of the disease.
In the 1970s and 1980s, it became clear that HIT antibodies activated both platelets and endothelial cells.3,4 Further analysis showed that blocking platelet FcγRIIA inhibited platelet activation by HIT sera in vitro,5 suggesting that platelet activation involved an immune complex. In the early 1990s, this complex was identified as heparin bound to the platelet-specific chemokine, platelet factor 4 (PF4).6 Over the past 20 years, additional insights into the mechanism(s) underlying this immune complex disorder have emerged that have advanced our understanding of why this disorder is particularly prothrombotic and occurs in a only a small subset of patients. Additionally, advances have been made on the clinical side with respect to prevention, diagnosis, and treatment.
EPIDEMIOLOGY
The frequency of HIT in heparin-treated patients ranges from less than 0.1 percent to 5.0 percent, depending on patient- and heparin-specific risk factors. These include the patient population, gender, nature of the heparin used, and duration of heparin exposure (Table 118–1).
| Patient-Specific Factors | Heparin-Specific Factors |
|---|---|
| Patient population (surgical > medical > obstetric > pediatric) | Type of heparin (unfractionated heparin > low-molecular-weight heparin) |
| Major trauma > minor trauma | Duration of heparin (~5 days >shorter courses) |
| Sex (female > male) |
The most important determinant of risk is the patient population. In a meta-analysis of seven prospective studies, the incidence of HIT was greater among surgical than medical patients (odds ratio [OR]: 3.25; 95 percent confidence interval [CI]: 1.98 to 5.35).7 The incidence of HIT approaches 5 percent in patients who receive unfractionated heparin (UFH) after major orthopedic surgery.8 Patients undergoing surgery with cardiopulmonary bypass have a very high frequency of anti-PF4/heparin antibody seroconversion (50 to 75 percent by postoperative day 10), but a lesser incidence of HIT (0.5 to 1.0 percent).8,9,10 HIT occurs in 0.5 to 1.0 percent of medical patients7 and in less than 0.1 percent of pregnant women11,12 and children.13 In a randomized trial of trauma patients, major trauma was associated with a significantly greater incidence of HIT than minor trauma (2.2 percent vs. 0.0 percent, p = 0.01) despite identical heparin exposure.14
Female sex is also a risk factor for HIT. A meta-analysis found an approximately twofold greater risk of HIT in women than men (OR: 2.37; 95 percent CI: 1.37 to 4.09). Analyses of a German database and a randomized trial of UFH versus low-molecular-weight heparin (LMWH) after orthopedic surgery yielded similar findings.7
HIT is more common with UFH than LMWH in surgical patients. In a meta-analysis of 15 studies, primarily involving orthopedic surgery patients, the incidence of HIT with UFH and LMWH was 2.6 percent and 0.2 percent, respectively.15 Data are conflicting on whether the risk of HIT is reduced with LMWH in medical patients.7,16,17 Fondaparinux, a synthetic pentasaccharide anticoagulant, is associated with a nearly negligible risk of HIT, although several cases of fondaparinux-associated HIT have been reported.18
Duration of heparin exposure also influences the risk of HIT. In a meta-analysis of 3529 patients receiving UFH thromboprophylaxis for 6 or more days, the incidence of HIT was 2.6 percent.15 Review of a hospital database indicated that briefer courses induce a substantially lower incidence of HIT (0.2 percent).19
High-quality data on the impact of dose and route of administration of heparin on the risk of HIT are lacking. Some studies suggest a lower rate of HIT with prophylactic dose subcutaneous UFH than therapeutic dose intravenous UFH,19 but these analyses are confounded by differences in the patients that receive these treatments including the clinical indication for heparin. Rarely, HIT has been reported with very low doses of heparin such as with use of heparin flushes or heparin-bonded catheters.20,21
ETIOLOGY AND PATHOGENESIS
The development of HIT antibodies is nonclassical in that these antibodies typically begin as IgG and not IgM,22 may disappear after a few months, and may not reappear with heparin reexposure.23 It has been proposed that the initial antigen exposure involves PF4 complexed with bacterial wall (Fig. 118–1) and that antibodies against this complex may be an important antimicrobial defensive mechanism.24 These antigenic complexes, as well as circulating PF4–heparin complexes, are detected by splenic marginal B cells that subsequently produce pathogenic HIT antibodies.25
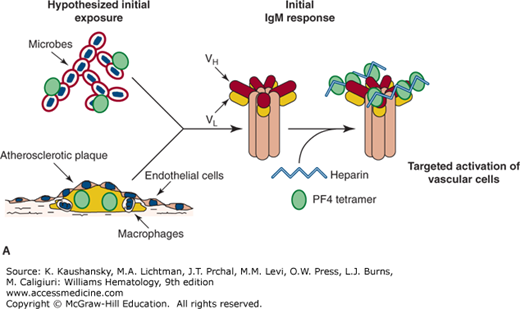
Figure 118–1.
Proposed etiology of the immune response in heparin-induced thrombocytopenia (HIT). A. Proposed first exposure to HIT antigenic complex either during microbial invasion24 or within growing atherosclerotic plaques, which are known to contain both platelet factor 4 (PF4) and immune-responsive cells.49 In both cases, soluble PF4 and negatively charged molecules must be presented to B cells and result in an initial immunoglobulin M—perhaps low affinity—response. B. On exposure to heparin, especially unfractionated heparin, complexes form with free PF4 and are presented to splenic marginal B cells,25 which subsequently produce pathogenic HIT antibodies that bind with high affinity to PF4 complexed to surface GAG complexes.29 The concentrated binding of HIT antibodies to surface PF4–GAG complexes may enhance FcγRIIA aggregation and subsequent platelet activation.
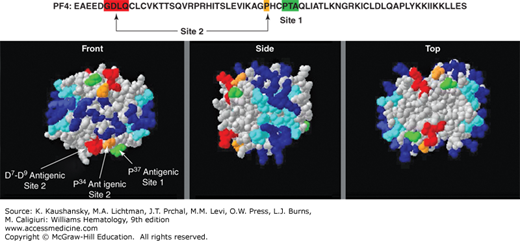
The nature of the antigenic heparin–PF4 complex has been partially defined. At the concentrations reached at sites of injury, PF4 exists as a tetramer. Crystal structure analysis shows that this tetramer is encircled by a ring of positive charge (Fig. 118–2),26 and heparin is thought to bind to this region.27 There are two closely spaced HIT antibody recognition domains (Fig. 118–2).28 These domains are distinct from the heparin-binding domain. About half of patients have antibodies that react with one or the other HIT antigenic domain and one-third of patients do not have antibodies that react to either domain, suggesting that there are other HIT antigenic sites on PF4. Studies of antihuman PF4 monoclonal antibodies suggest that unlike nonpathogenic anti-PF4 antibodies, HIT antibodies markedly increase their binding affinity for PF4 maintained in a tetrameric as opposed to dimeric state.29 Moreover, tetrameric PF4 and UFH need to be at approximately 1:1 molar ratio for optimal HIT antigenicity.30,31 At this ratio, ultralarge complexes (>670 kDa) of PF4 and heparin form that appear as visible colloidal complexes, and these are likely to be the antigenic source in HIT.32,33 At higher or lower ratios, PF4 predominantly forms smaller and less antigenic PF4–heparin complexes. Ultralarge complexes are inefficiently formed with LMWH, offering a potential explanation as to why this class of agents is associated with a lower incidence of HIT compared with UFH. Fondaparinux does not form large complexes with PF4, explaining the negligible incidence of HIT with this agent and supporting its potential utility in the prevention or treatment of HIT.
Figure 118–2.
Platelet factor 4 (PF4) tetramer structure. At the top is the linear sequence of PF4. The regions that are known to contribute to heparin-induced thrombocytopenia (HIT) antigenicity when PF4 is complexed to heparin are boxed. Below are three views of the PF4 tetramer with the positively charged residues shown in both light and dark blue. Sites at which HIT neoepitopes are exposed on the PF4 tetramer are indicated. (Adapted with permission from Li ZQ, Liu W, Park KS, et al: Defining a second epitope for heparin-induced thrombocytopenia antibodies using KKO, a murine HIT-like monoclonal antibody. Blood 99(4):1230–1236, 2002.)
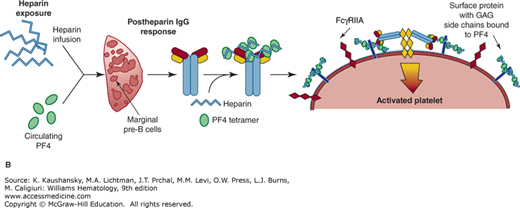
A passive immunization murine model of HIT has been used to demonstrate the following components are necessary to induce both thrombocytopenia and a prothrombotic state in HIT: the presence of human PF4 in platelets, FcγRIIA on the surface of platelets and possibly other vascular cells, and the presence of a pathogenic HIT-like antibody.34 Heparin has a more complex relationship to the pathogenesis of HIT (Fig. 118–3). In the passive immunization model of HIT, wherein a pathogenic HIT-like antibody is infused into mice expressing both human PF4 and FcγRIIA, heparin infusion is not needed to cause thrombocytopenia and a prothrombotic state.35 The explanation for a lack of need to infuse heparin in this model is as follows: One important potential role of heparin in the pathogenesis of HIT is to form soluble, circulating PF4–heparin complexes that induce anti-PF4–heparin antibody production by presentation of the complex to splenic marginal B cells (see Figs. 118–1 and 118–3).25 Passive immunization with HIT antibodies in these mice obviates the need for delivery of soluble antigenic complexes to the spleen. Another role for infused heparin is counterintuitive in that it may prevent HIT by partially or completely removing surface-bound PF4 (Fig. 118–3).35 If the level of circulating, free human PF4 in the mice was initially low relative to the level of infused heparin, all surface-bound PF4 and detectable surface antigenicity would be removed and the circulating HIT antibodies would have no targets on platelets and other vascular cells (Fig. 118–3). HIT therefore would not develop in spite of the presence of heparin and circulating HIT antibody. On the other hand, if the level of circulating free human PF4 in the mice was initially high relative to the infused heparin, not all surface-bound PF4 would be removed. Circulating HIT antibodies could then target and activate platelets and other vascular cells, leading to thrombocytopenia and thrombosis (Fig. 118–3).
Figure 118–3.
The role(s) of heparin in heparin-induced thrombocytopenia (HIT). On the left is depicted a quiescent platelet surface with GAG-expressing proteins as well as individual FcγRIIA receptors. Platelet factor 4 (PF4) is normally released by platelets in the steady-state, especially in individuals with underlying inflammation and/or atherosclerosis. Additionally, individuals have a wide range of platelet PF4 content, and this, too, contributes to having individuals with different levels of surface PF4 binding. Heparin infusions leads to HIT immunoglobulin G formation (see Fig. 118-1B), but also removes surface-bound PF4. If the individual has little initial surface-bound PF4, the surface of the platelets will be wiped clean of bound PF4 by the infused heparin and not be targeted by the HIT antibodies (right, top) so that HIT antibodies circulate but HIT does not develop. If there is significant residual surface-bound PF4 after heparin infusion, HIT antibodies will attach to the cell surface and activate the platelets (right, bottom), potentially leading to HIT.
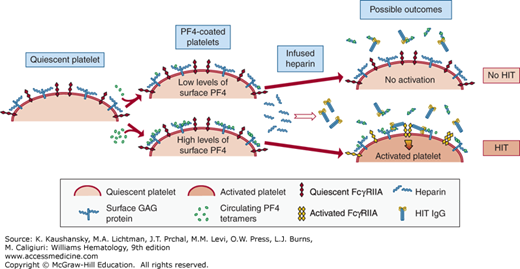
Most patients likely begin with little PF4 bound to surface glycosaminoglycans (GAGs). After therapeutic heparinization, the level of surface PF4 goes down markedly so that the platelets cannot be targeted by anti-PF4–heparin HIT antibodies. However, patients with high levels of surface PF4 and significant surface PF4 antigenic complexes remaining after heparinization may be at risk for binding of pathogenic anti-PF4–heparin HIT antibodies to platelets and other vascular cells. Bound pathogenic antibodies lead to thrombocytopenia by clearance of antibody-coated platelets by the reticuloendothelial system. Bound antibodies also lead to platelet activation through FcγRIIA and the formation of procoagulant platelet microparticles that contribute to thrombosis.36
As part of this activation, HIT antibodies also bind to endothelial cells likely via PF4–surface GAG complexes,4,37 leading to local vascular activation and contributing to further local thrombosis. Additionally, HIT antibodies activate PF4-targeted monocytes38,39 and neutrophils.40 Monocyte activation may involve its surface Fcγ receptors41 with subsequent increased tissue factor expression and other changes consistent with a prothrombotic and inflammatory state. Monocyte and neutrophil GAGs are more complex than GAGs on the surface of platelets, which are mostly chondroitin sulfate42 and have relatively low affinity for PF4.43 Monocytes and neutrophils bind PF4 with greater avidity and are more resistant to removal of bound PF4 by circulating heparin than platelets. In HIT, they may be preferentially targeted and activated relative to the platelets, contributing to the prothrombotic state of this immune thrombocytopenia.44
Are there any genetic polymorphisms that are associated with an increased risk of developing HIT or developing thrombosis after HIT begins? No clear linkage has been shown with known thrombophilic polymorphisms including Factor VLeiden, ProthrombinG20210A, MTHFRC677T, αIIbβ3 and α1β2.45 Studies addressing a functional FcγRIIAR/H131 polymorphism had varied outcome.46,47 It is unclear whether patients with HIT have a higher density of FcγRIIA on their platelets.48 High IgG affinity for the heparin–PF4 complex appears to affect the risk of developing HIT.
The model shown in Fig. 118–3 suggests that individuals with high PF4 content in their platelets and/or sustained platelet activation as might be seen in patients with significant atherosclerosis, a postsurgery state, or trauma would be most likely to develop HIT after heparinization and HIT antibody development. However, a relationship between formation of HIT antibodies and the degree of atherosclerosis in patients undergoing cardiopulmonary bypass surgery was not noted.49 If levels of PF4 on the surface of circulating cells determine risk of developing HIT, this would offer a potential method for prescreening patients prior to heparinization and eliminate those at increased risk of HIT or as a useful tool in heparinized patients who develop thrombocytopenia to see whether they potentially can develop HIT. Theoretically, only those heparinized individuals with detectable surface PF4 would be potential candidates for developing HIT if they concurrently develop pathogenic antibodies.
CLINICAL DIAGNOSIS
The clinical hallmark of HIT is development of thrombocytopenia in the setting of a proximate heparin exposure. The combination of thrombocytopenia and heparin exposure in hospitalized patients is common and has poor specificity for HIT.50 Therefore, other clinical clues must be sought in estimating the clinical likelihood of HIT. These include timing, degree of platelet count fall, nadir platelet count, presence of thromboembolism or hemorrhage, and the likelihood of other causes of thrombocytopenia.
The platelet count in HIT characteristically begins to fall 5 to 10 days after initial heparin exposure.23 There are three exceptions to this rule: (1) in rapid-onset HIT, patients with recent heparin exposure (within the previous 90 days) and preformed anti-PF4–heparin IgG experience a fall in platelet count immediately upon reexposure; (2) in delayed-onset HIT, clinical manifestations develop a median of 10 to 14 days after heparin is discontinued51,52; and (3) a small number of patients with spontaneous HIT have been reported. These patients present with a thrombotic thrombocytopenic disorder reminiscent of HIT in the absence of recognized heparin exposure.53 Both delayed-onset HIT and spontaneous HIT occur in the absence of circulating heparin and may involve pathogenic HIT antibodies that recognize complexes of PF4 and endogenous GAGs on blood and vascular cells.
The percentage fall in platelet count is measured from the peak platelet count after initiation of heparin to the nadir platelet count. Most patients with HIT experience a 50 percent or greater fall in platelet count; a more modest decline (30 to 50 percent) occurs in approximately 10 percent of patients.54
As opposed to most other forms of drug-induced immune thrombocytopenia, thrombocytopenia associated with HIT is characteristically mild or moderate. The median nadir platelet count is approximately 60 × 109/L and rarely falls below 20 × 109/L in the absence of concomitant disseminated intravascular coagulation (DIC).54 The nadir platelet count in HIT need not meet the traditional definition of thrombocytopenia (<150 × 109/L). For example, patients with postoperative thrombocytosis may experience a subsequent greater than 50 percent decline in platelet count attributable to HIT that does not fall below this threshold.55
Thromboembolism is the presenting feature in up to 25 percent of patients with HIT and complicates approximately half of all cases.56,57 Lower-extremity deep vein thrombosis and pulmonary embolism are the most common thrombotic manifestations, outnumbering arterial events by approximately 2:1.56 Major venous obstruction can lead to limb gangrene. Catheter-associated upper extremity deep venous thrombosis is common.58 Arterial thromboembolism most frequently involves the extremities, but may also manifest as stroke or myocardial infarction.56 Thrombosis of other vascular beds including cerebral sinuses, mesenteric vessels, and adrenal veins is well-documented as is thrombotic occlusion of vascular grafts, fistulas, and extracorporeal circuitry.
In contrast to most other forms of drug-induced immune thrombocytopenia, spontaneous hemorrhage is rare in HIT, even when thrombocytopenia is severe. In a prospective study, bleeding complications were not increased in HIT patients compared with nonthrombocytopenic controls.59
Rare sequelae of HIT include anaphylactoid reactions after intravenous heparin bolus, transient global amnesia, and skin necrosis at subcutaneous heparin injection sites.60,61 Curiously, these phenomena may occur in the absence of thrombocytopenia. Nonnecrotizing erythematous injection site lesions are generally caused by delayed type IV hypersensitivity rather than HIT.62
The likelihood of other etiologies of thrombocytopenia must be carefully considered in patients with suspected HIT. Common causes of hospital-acquired thrombocytopenia include infection; drugs other than heparin; DIC; dilution; and intravascular devices and extracorporeal circuits such as intraaortic balloon pumps, cardiopulmonary bypass, and extracorporeal membrane oxygenation.63
Clinical scoring systems have been developed to permit estimation of the probability of HIT based on the aforementioned features. The most extensively studied of these systems, the 4T score,64 classifies the probability of HIT as low, intermediate, or high on the basis of four criteria: Thrombocytopenia, Timing, Thrombosis or other sequelae, and the likelihood of other causes of thrombocytopenia (Table 118–2). In a meta-analysis of 13 studies, the negative predictive value of a low probability 4T score was 99.8 percent (95 percent CI: 97.0 to 100.0). The positive predictive value of an intermediate and high probability 4T score was 14 percent (95 percent CI: 9 to 22) and 64 percent (95 percent CI: 40 to 82), respectively.65 The 4T score is limited by moderate interobserver agreement.66 An alternative scoring system, the HIT Expert Probability (HEP) Score, exhibited improved reliability and favorable operating characteristics in a retrospective study, but remains to be prospectively validated.67
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


