Heparin and Vascular Proteoglycans
Douglas M. Tollefsen
Lijuan Zhang
Heparin is a sulfated glycosaminoglycan isolated from mammalian tissues that are rich in mast cells. When administered intravenously, heparin binds to the plasma protein antithrombin (previously known as antithrombin III), causing it to rapidly inhibit proteases of the intrinsic and common coagulation pathways. This interaction produces a potent anticoagulant effect. Endogenous heparin-like molecules appear to be involved in the inhibition of coagulation within normal blood vessels and may have a variety of other biologic functions. Although heparin is an effective agent for the treatment of venous thromboembolic disease, unstable angina, and acute myocardial infarction, it occasionally produces lifethreatening bleeding or triggers an immune reaction, causing thrombocytopenia associated with venous and arterial thrombosis.
HISTORICAL OVERVIEW
The term heparin was used by Howell1 in 1923 to describe an aqueous extract of canine liver that inhibited coagulation of blood in vitro. These extracts later were shown to consist of mixtures of sulfated polysaccharides containing uronic acid and glucosamine.2 The suggestion that heparin might be used to treat thromboembolism was initially greeted with skepticism because of the fear that patients given heparin would bleed to death. According to Jaques,3 this fear was allayed by a dramatic demonstration conducted by Charles Best at the University of Toronto in 1937:
Best asked me [Jaques] to anesthetize a dog, prepare it for laparotomy, and inject heparin intravenously. When the most illustrious surgeons in the world arrived, Best explained what I had done (a blood sample which I had taken was incoagulable). Best then picked up a scalpel and handed it to Dr. Balfour with the request that he make a midline incision. The surgeons, properly dressed in their morning suits, jumped back, whereupon Best made the incision and I applied haemostats to two tiny bleeding points and showed a clean dry incision. Within months of this demonstration, surgeons all over the world took up heparin enthusiastically.
Canadian and Swedish investigators reported success in the use of heparin to treat recurrent thrombosis and pulmonary embolism in 1939.2,4 The indications for heparin were soon expanded to include vascular surgical procedures, extracorporeal circulation, and prophylaxis of thromboembolism.
Despite the ensuing widespread clinical use of heparin, its mechanism of action remained obscure for several decades. As early as 1895, “thrombin” had been observed to lose activity gradually when added to defibrinated plasma or serum5; this inhibitory potential became known as the progressive antithrombin activity. In 1939, Brinkhous et al.6 showed that heparin was effective as an anticoagulant only in the presence of a nondialyzable plasma component that they termed heparin cofactor. These early observations were linked in 1968, when Abildgaard7 reported that a single protein (i.e., antithrombin) isolated from human plasma possessed both progressive antithrombin and heparin cofactor activity. Rosenberg and Damus8 purified larger quantities of antithrombin, enabling a more detailed physicochemical characterization of the protein. Subsequently, a second protein with heparin cofactor activity was purified from plasma by Tollefsen et al.9 and termed heparin cofactor II (HCII).
In the sections that follow, we discuss the structure, biosynthesis, and isolation of heparin and heparan sulfate (HS); the synthesis of anticoagulantly active heparan sulfate proteoglycans (HSPGs) by endothelial cells; the physiologic role of the heparin-antithrombin system; some of the nonanticoagulant actions of heparin; the synthesis of anticoagulant heparin analogs; and the interaction of HCII with glycosaminoglycans. A discussion of the structures and mechanisms of action of antithrombin and HCII is presented in Chapter 18.
STRUCTURE AND BIOSYNTHESIS OF HEPARIN AND HEPARAN SULFATE
Heparin is a highly sulfated polymer built on a backbone of alternating D-glucuronic acid and N-acetyl-D-glucosamine residues10,11 that is widely distributed in the animal kingdom, being found in both vertebrates and invertebrates.12 In mammals, heparin is initially synthesized in mast cells as a heparin proteoglycan in which multiple heparin chains, each containing 100 to 150 disaccharide units, are covalently linked to a core protein called serglycin.13,14 Once generated, the heparin proteoglycan is degraded by three different types of lysosomal enzymes: (i) proteases that cleave the polypeptide chain; (ii) endoglycosidases that cut the heparin chain between glucuronic acid and glucosamine residues; and (iii) exoglycosidases that remove monosaccharide units from the nonreducing ends of heparin oligosaccharides.15 The degraded products are then stored in the secretory granules of mast cells, which are widely distributed in a variety of organs, including the liver, heart, lungs, kidneys, and intestine.
Mammalian cells also synthesize HSPGs that consist of core proteins with covalently attached HS chains of 50 to 150 disaccharide units.16 Both heparin and HS chains exhibit structural diversity, which arises from various patterns of N-sulfation, N-acetylation, and O-sulfation of the disaccharide units. The distinction between heparin proteoglycan and HSPGs is based on (i) the structure of the core protein; (ii)
the location of the glycoconjugate within the cell; and (iii) the relative amounts of N-acetylglucosamine and glucuronic acid present in the carbohydrate chains. In particular, the heparin core protein is small and relatively simple in structure with extended runs of Ser-Gly attachment sites,13,14,17 whereas HS core proteins are large and more diverse in structure18 with short runs of Ser-Gly or closely spaced Ser-Gly attachment sites19; heparin proteoglycan is found within mast cell granules, whereas HSPGs are located on cell surfaces or in the surrounding matrix20; and heparin contains larger amounts of N-sulfate, O-sulfate, and iduronic acid, whereas HS is less extensively modified.
the location of the glycoconjugate within the cell; and (iii) the relative amounts of N-acetylglucosamine and glucuronic acid present in the carbohydrate chains. In particular, the heparin core protein is small and relatively simple in structure with extended runs of Ser-Gly attachment sites,13,14,17 whereas HS core proteins are large and more diverse in structure18 with short runs of Ser-Gly or closely spaced Ser-Gly attachment sites19; heparin proteoglycan is found within mast cell granules, whereas HSPGs are located on cell surfaces or in the surrounding matrix20; and heparin contains larger amounts of N-sulfate, O-sulfate, and iduronic acid, whereas HS is less extensively modified.
Biosynthesis of a heparin or HS proteoglycan occurs in the Golgi apparatus and is initiated by attachment of a linkage region tetrasaccharide containing one xylose, two galactose, and one glucuronic acid to the serine hydroxyl group of a Ser-Gly (or Ala) dipeptide in the core protein.10,11 The formation of heparin/HS chains depends on nearby acidic or hydrophobic residues or close spacing of Ser-Gly sites.19,20,21 In humans, the heparin core protein serglycin contains an extended run of eight Ser-Gly repeats, in which virtually all of the Ser residues become modified with the linkage region tetrasaccharide. The tetrasaccharide is synthesized by the sequential action of four glycosyltransferases (xylosyltransferase, galactosyltransferase I, galactosyltransferase II, and glucuronyltransferase I) that use uridine diphosphate (UDP) sugars as their substrates. The structures of serglycin and its linkage region are depicted in FIGURE 45.1.
Immediately after formation of the linkage region tetrasaccharide, α–N-acetylglucosaminyltransferase I adds an N-acetylglucosamine residue and thereby commits the nascent chain to the heparin/HS biosynthetic pathway (see FIGURE 45.2).11 Linkage region tetrasaccharides on other core proteins can be modified at this stage by β–N-acetylgalactosaminyltransferase I, resulting in biosynthesis of chondroitin sulfate or dermatan sulfate chains.20 The heparin/HS chain is assembled by the alternate incorporation of glucuronic acid and N-acetylglucosamine from the corresponding UDP sugars. This is accomplished through the action of copolymerases that are members of the EXT gene family having β-glucosaminyltransferase II and α–N acetylglucosaminyltransferase II activities. Thereafter, this simple copolymer is modified by a concerted process that leads to different modifications in various regions of the glycosaminoglycan chain.
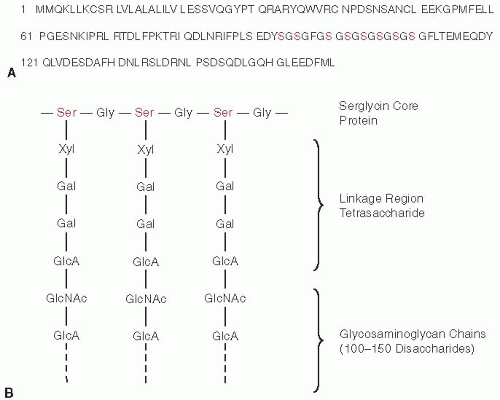 FIGURE 45.1 Heparin proteoglycan structure. A: Amino acid sequence of the human serglycin core protein. The red-colored residues comprise the glycosaminoglycan attachment sites. B: Structure of the linkage region. Ser, serine; Gly, glycine; Xyl, xylose; Gal, galactose; GlcA, glucuronic acid; GlcNAc, N-acetyl-glucosamine. |
First, glucosamine residues are partially N-deacetylated, and the exposed amino groups serve as acceptors of sulfate groups in a transfer reaction with 3′-phosphoadenosine-5′-phosphosulfate (PAPS). These two reactions are carried out by a bifunctional enzyme, N-acetylglucosamine N-deacetylase/N-sulfotransferase (NDST), which is composed of a single polypeptide chain. Four isoforms of human NDST (NDST-1, -2, -3, and -4) have been identified, differing in their expression in various tissues and in their relative N-deacetylase and N-sulfotransferase activities.22 Mice that lack NDST-2 do not synthesize mast cell heparin but produce HS,23,24 whereas mice that lack NDST-1 produce undersulfated HS and die at birth of lung failure.25,26 At this stage of biosynthesis, the nascent heparin or HS polymer contains stretches of N-sulfoglucosamine interspersed with unmodified N-acetylglucosamine residues.
Next, glucuronic acid residues are variably epimerized to iduronic acid by glucuronyl C5-epimerase. Then the iduronic acid residues and, to a lesser extent, glucuronic acid residues are partially O-sulfated at the C2-position. Finally, the glucosamine moieties are O-sulfated to a variable extent at the C6- and the C3-positions. Epimerization and O-sulfation occur only in regions of the polymer that contain
N-sulfoglucosamine, but these modifications are incomplete. As a result, a wide variety of oligosaccharide structures can be formed, each having the ability to interact with a different protein (e.g., antithrombin) or class of proteins (e.g., fibroblast growth factors [FGFs] or viral capsid proteins).10 One HS 2-O-sulfotransferase, three HS 6-O-sulfotransferases, and six HS 3-O-sulfotransferases have been identified in humans.22 Multiple isoforms make possible tissue-specific and developmentally regulated expression of the corresponding enzyme activities. Because the enzymatic properties of the isoforms may differ in subtle ways, their selective expression may be responsible for unique oligosaccharide sequences found in heparin/HS in different tissues and different animals. For example, 3-O-sulfotransferase-1 (3-OST-1) is primarily responsible for sulfation of the C3-position of glucosamine in the sequence GlcA-GlcNS, which gives rise to the highaffinity antithrombin-binding site (discussed in the subsequent text).
N-sulfoglucosamine, but these modifications are incomplete. As a result, a wide variety of oligosaccharide structures can be formed, each having the ability to interact with a different protein (e.g., antithrombin) or class of proteins (e.g., fibroblast growth factors [FGFs] or viral capsid proteins).10 One HS 2-O-sulfotransferase, three HS 6-O-sulfotransferases, and six HS 3-O-sulfotransferases have been identified in humans.22 Multiple isoforms make possible tissue-specific and developmentally regulated expression of the corresponding enzyme activities. Because the enzymatic properties of the isoforms may differ in subtle ways, their selective expression may be responsible for unique oligosaccharide sequences found in heparin/HS in different tissues and different animals. For example, 3-O-sulfotransferase-1 (3-OST-1) is primarily responsible for sulfation of the C3-position of glucosamine in the sequence GlcA-GlcNS, which gives rise to the highaffinity antithrombin-binding site (discussed in the subsequent text).
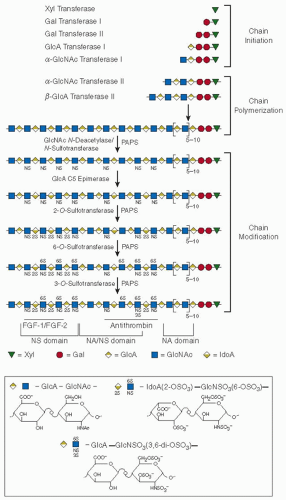 FIGURE 45.2 Heparin/HS biosynthesis. The symbols are defined by the structures shown below the scheme. Representative tracts of contiguous N-acetylated disaccharide units (NA domains), contiguous N-sulfated sequences of variable length (NS domains), and alternating N-acetylated and N-sulfated units (NA/NS domains) in HS are indicated. Heparin is predominantly composed of a single, extended NS domain. Regions that have been implicated in binding of fibroblast growth factors FGF-1/FGF-2 and antithrombin are shown. IdoA, iduronic acid; PAPS, 3′-phosphoadenosine-5′-phosphosulfate. Other abbreviations are defined in the legend to FIGURE 45.1. (Reprinted from Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest 2001;108:169-173, with permission.) |
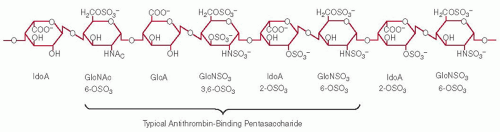 FIGURE 45.3 Representative region of a heparin chain containing a typical antithrombin-binding sequence. IdoA, iduronic acid; GlcA, glucuronic acid; GlcNAc, N-acetyl-glucosamine. |
ANTITHROMBIN-BINDING SEQUENCES IN HEPARIN
A segment of a heparin chain containing an antithrombin-binding site is depicted in FIGURE 45.3. It is important to emphasize that this sequence is only one of many possible arrangements of uronic acid and glucosamine moieties found elsewhere in the polymer. As progress was being made in defining various aspects of the biosynthesis, linear sequence, and three-dimensional configuration of heparin and HS, it was tacitly assumed that heparin may exhibit numerous alternative structures equally capable of interacting with antithrombin.27 This assumption was shown to be incorrect in 1976, when several groups demonstrated that only approximately 30% of commercial heparin is able to bind to antithrombin but that the bound fraction is responsible for virtually all of the anticoagulant activity.28,29,30
These findings led to investigations designed to determine the structural differences between active/high-affinity (AT+) and in active/low-affinity (AT-) heparin species.27 Rosenberg et al.31 showed that oligosaccharides produced from high-affinity heparin were enriched in a tetrasaccharide sequence that contained glucuronic acid and N-acetylglucosamine. Leder32 isolated an enzyme that removes sulfate from the 3-O position of glucosamine, and Lindahl et al.33 showed that this substituent is critical for high-affinity binding of heparin to antithrombin. These observations were confirmed and extended by Choay et al.34,35 who synthesized a series of pentasaccharides that are able to catalyze the factor Xa-antithrombin reaction. The contributions of individual substituents within the antithrombin-binding pentasaccharide have been evaluated by comparing the avidity of synthetic oligosaccharides for antithrombin.36 On the basis of these data, the relative importance of individual residues is shown in FIGURE 45.4. The presence of an N-sulfate in place of the N-acetyl group on the first glucosamine residue increases the affinity for antithrombin approximately twofold. Similarly, the 2-O-sulfate and 6-O-sulfate groups on the fourth and fifth residues of the pentasaccharide increase the affinity but are not
absolutely required for high-affinity binding. Therefore, some variability in the structure of heparin at these sites is compatible with antithrombin binding and anticoagulant activity. In contrast, certain structures are critical for antithrombin binding, including the presence and orientation of carboxylate groups on the two uronic acid residues, the 6-O-sulfate on the first glucosamine, the 3-O-sulfate and N-sulfate on the middle glucosamine, and the N-sulfate on the last glucosamine of the pentasaccharide.
absolutely required for high-affinity binding. Therefore, some variability in the structure of heparin at these sites is compatible with antithrombin binding and anticoagulant activity. In contrast, certain structures are critical for antithrombin binding, including the presence and orientation of carboxylate groups on the two uronic acid residues, the 6-O-sulfate on the first glucosamine, the 3-O-sulfate and N-sulfate on the middle glucosamine, and the N-sulfate on the last glucosamine of the pentasaccharide.
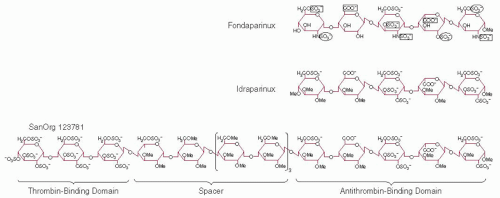 FIGURE 45.4 Synthetic heparin-like oligosaccharides. Sulfate and carboxylate groups required for high-affinity binding to antithrombin are indicated by boxes in the structure of fondaparinux. Sulfate groups that are not essential but that contribute to high-affinity binding are circled. |
Although synthetic heparin pentasaccharides that bind to antithrombin with high affinity accelerate the factor Xa-antithrombin interaction, they do not significantly increase the rate of neutralization of thrombin or other hemostatic enzymes. Isolation of oligosaccharides derived from partial chemical depolymerization of heparin revealed that molecules at least 16 monosaccharide units in length are required to accelerate both the thrombin-antithrombin and factor Xa-antithrombin interactions.37 For inhibition of factor Xa to occur, it is sufficient to form a binary complex between heparin and antithrombin that leads to a conformational change in the reactive site loop of the inhibitor. To facilitate thrombin inhibition, however, longer heparin chains must bind simultaneously to thrombin and antithrombin to form a ternary thrombin-heparin-antithrombin complex. These differences in mechanism of inhibition explain why smaller heparin molecules can inhibit factor Xa while failing to inhibit thrombin. The inhibitory mechanism of antithrombin is discussed in more detail in Chapter 18.
SYNTHESIS OF ANTICOAGULANTLY ACTIVE HEPARAN SULFATE PROTEOGLYCANS BY ENDOTHELIAL CELLS
Mast cells were once thought to be the only site of synthesis of anticoagulantly active heparin. These cells are located beneath the endothelium and sequester heparin within their secretory granules,38 where it forms crystalline arrays with cationic molecules such as histamine and neutral proteases. Ordinarily, heparin is not released from mast cells into the circulation; one exception may be patients with systemic mastocytosis, in whom small amounts of heparin can appear in the bloodstream and slightly prolong the activated partial thromboplastin time.39 Therefore, mast cell heparin is unlikely to affect hemostasis under normal circumstances. These considerations led to the suggestion that endothelial cells might synthesize HSPGs endowed with the critical oligosaccharide structure required to bind and activate antithrombin.40 In parallel investigations of the structure of anticoagulantly active mast cell heparin, HSPGs synthesized by endothelial cells were shown to contain a small subpopulation (˜1%) of HS that binds antithrombin with high affinity (HSAT+) and accelerates inhibition of thrombin and factor Xa by antithrombin in vitro.41 These studies suggested that HSAT+ might be a major physiologic modulator of hemostasis.
Early investigations revealed that HS is present in the aorta and other vessels and exhibits trace amounts of anticoagulant activity.42,43 It proved difficult, however, to rule out the possibility that mast cells were the source of this material. Direct evidence that endothelial cells are able to generate anticoagulantly active HSPGs was eventually obtained by Marcum and Rosenberg,44 who studied cloned microvascular endothelial cells isolated from the rat epididymal fat pad. They showed that polysaccharides extracted from these cells possessed antithrombin accelerating activity that could be completely eliminated by treatment with purified Flavobacterium heparinase. Virtually all of the heparin-like material could be harvested by a brief exposure of the endothelial cells to dilute trypsin, which suggested that these components are located on the cell surface. Furthermore, metabolic labeling of the polysaccharides with [35S]sulfate, followed by affinity fractionation with antithrombin and structural analysis, revealed the presence of oligosaccharide structures typically found in anticoagulantly active commercial heparin. Similar studies conducted with cloned bovine aortic endothelial cells gave virtually identical results and indicated that the level of anticoagulantly active HS species present would correspond to approximately 50,000 antithrombin-binding sites per endothelial cell.45 This estimate of surface-bound anticoagulantly active polysaccharide was in excellent agreement with the data generated by characterization of the interactions of antithrombin with bovine aortic tissue sections.
Two major endothelial cell core proteins that are endowed with anticoagulantly active HS chains have been characterized. Kojima et al.46 isolated intact HSPGs from rat microvascular endothelial cells using a combination of ion-exchange chromatography, affinity fractionation with antithrombin, and gel filtration in denaturing solvents. The anticoagulantly active HSPGs bound to antithrombin (HSPGAT+) constituted approximately 5% of the total HSPGs and were endowed almost entirely with anticoagulantly active HS chains, whereas the anticoagulantly inactive HSPGs that did not interact with antithrombin (HSPGAT-) represented approximately 95% of the total HSPGs.
The two types of HS chains had the same molecular mass of approximately 25 to 30 kDa, and the anticoagulantly active HS chains possessed a single antithrombin-binding region. The core proteins of HSPGAT+ and HSPGAT- were isolated after treatment with Flavobacterium heparitinase and purification by ionexchange chromatography. Peptide mapping studies suggested that HSPGAT+ and HSPGAT- contained the same two major core proteins with molecular masses of 50 and 30 kDa. The primary sequences of internal peptides derived from the HSPG core proteins were used to clone two cDNAs from a rat microvascular endothelial cell library.47 The first cDNA constituted a previously unidentified species, termed syndecan-4 (ryudocan), whereas the second cDNA represented the rat ortholog of syndecan-1. The latter molecule was a known proteoglycan that was originally thought to be limited in its distribution to epithelial cells. The two cDNAs encode integral membrane proteins of 202 amino acids (syndecan-4) and 313 amino acids (syndecan-1), which have extraordinarily homologous transmembrane and intracellular domains but very distinct extracellular regions. Syndecan-4 has three potential glycosaminoglycan attachment sites in its extracellular domain, whereas syndecan-1 has five. Both molecules are expressed at high levels (0.1% to 0.5% of total mRNA) in primary endothelial cells, primary smooth muscle cells, and primary fibroblasts. The possibility that slightly different core proteins direct the synthesis of HSPGAT+ and HSPGAT- was excluded by stably expressing an epitope-tagged syndecan-4 in cells that synthesize anticoagulantly active and anticoagulantly inactive HS chains; after isolation of the proteoglycan with a specific antibody against the epitope, both HSAT+ and HSAT- were found to be present.48 This finding suggested that the presence of HS biosynthetic enzymes rather than expression of specific core proteins determines the fine structure of the HS chains. Further studies
suggested that in addition to syndecan-1 and syndecan-4, glypican-149 and perlecan50 synthesized by endothelial cells also bear anticoagulantly active HS chains.
suggested that in addition to syndecan-1 and syndecan-4, glypican-149 and perlecan50 synthesized by endothelial cells also bear anticoagulantly active HS chains.
The HS biosynthetic pathway generates limiting amounts of HSPGAT+ with regions of defined structure that contain the antithrombin-binding site; this pathway also produces the more abundant HSPGAT- with regions of varying structure that carry out other biologic functions. Analyses of cell mutants, created by overexpression of the syndecan-4 core protein or by chemical mutagenesis, revealed that anticoagulant HS generation requires a component present in limiting amounts.51,52 To identify this component, a cell-free system was developed by Shworak et al.53 in which wild-type microsomes were incubated with radiolabeled HSPG precursor extracted from mutant cells blocked during HSPGAT+ generation; coincubation of these two extracts resulted in the production of large amounts of HSPGAT+. Furthermore, in many different cell types, the concentration of this “microsomal conversion activity” correlated with the cell’s ability to generate HSPGAT+. Treatment of microsomal and cell surface HSPGs from wild-type cells with excess microsomal conversion activity transformed up to 35% of total HSPGs into HSPGAT+. This extent of conversion was much greater than the normal levels of cell surface HSPGAT+, which average approximately 1% to 5% of total HSPGs.53 These investigations provided evidence that a microsomal component limits transformation of small amounts of a HSPG precursor population into HSPGAT+.
The assay described in the preceding text was then used to isolate the limiting component, which proved to be the longsought glucosaminyl 3-OST-1.54,55 This enzyme usually is present in trace amounts as compared to other HS biosynthetic enzymes. When 3-OST-1 is no longer limiting, the cell’s capacity to generate HSAT+ is determined by the abundance of HSAT+ precursors.56 In vitro 3-O-sulfation with purified 3-OST-1 was used to tag the regions of the HSAT+ precursors destined to become antithrombin-binding sites and allow the HSAT+ precursors to be captured. The tagged regions were then structurally examined.57 It was shown that six 3-O-sulfation sites existed per HSAT+ precursor chain. At least five out of the six 3-O-sulfate-tagged oligosaccharides in HSAT+ precursors bound antithrombin, whereas none of 3-O-sulfate-tagged oligosaccharides from HSAT- precursors bound antithrombin. When treated with low pH nitrous acid or heparitinase, 3-O-sulfate-tagged HSAT+ and HSAT- precursors exhibited clearly different structural features: They had different uronic acid epimerization and O-sulfation patterns around the 3-O-sulfate acceptor sites. 3-O-sulfate-tagged HSAT+ hexasaccharides were antithrombin affinity purified, sequenced by chemical and enzymatic degradation, and shown to have structures compatible with the antithrombin-binding sequence illustrated in FIGURE 45.3. The differences in structure surrounding potential 3-O-sulfate acceptor sites in HSAT+ and HSAT- precursors suggested that these precursors might be generated by different concerted assembly mechanisms in the same cell.57
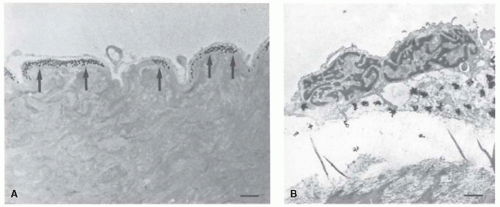 FIGURE 45.5 Autoradiography of normal rat aortas perfused with 125I-antithrombin. A: Light microscopic autoradiograph. Intense labeling is present in the basement membrane beneath the endothelial cells (arrows). Bar, 10 µm. B: Electron microscopic autoradiograph. Labeling is again evident beneath the endothelium. Bar, 1.0 µm. (Reprinted from de Agostini AI, Watkins SC, Slayter HS, et al. Localization of anticoagulantly active heparan sulfate proteoglycans in vascular endothelium: antithrombin binding on cultured endothelial cells and perfused rat aorta. J Cell Biol 1990;111:1293-1304, with permission.) |
ANATOMIC LOCATION OF ANTICOAGULANTLY ACTIVE HEPARAN SULFATE PROTEOGLYCANS
The anatomic location of HSPGAT+ was investigated by de Agostini et al.,58 who perfused normal rat aorta for short periods of time with 125I-antithrombin and observed that the labeled protein was bound to the aortic subendothelium (see FIGURE 45.5). Quantitation of the data revealed that only approximately 1% of the bound 125I-antithrombin was associated with the luminal surface of the endothelium and that only small amounts were associated with smooth muscle cells or connective tissue deeper in the vessel wall. The labeled antithrombin was also noted to bind to regions around capillaries, especially in the subendothelium associated with the basal lamina/basement membrane. Perfusion of the aorta with 125I-antithrombin-labeled
bovine serum albumin produced no detectable binding, which indicated that the binding of 125I-antithrombin within the aorta was not due to nonspecific interactions or trapping of the labeled protein. To demonstrate that the 125I-antithrombin specifically interacted with HS chains, the vasculature was perfused with Flavobacterium heparinase before perfusion with labeled antithrombin. This treatment effectively prevented any binding of 125I-antithrombin to all regions, including the subendothelium, while leaving the endothelial layer intact.
bovine serum albumin produced no detectable binding, which indicated that the binding of 125I-antithrombin within the aorta was not due to nonspecific interactions or trapping of the labeled protein. To demonstrate that the 125I-antithrombin specifically interacted with HS chains, the vasculature was perfused with Flavobacterium heparinase before perfusion with labeled antithrombin. This treatment effectively prevented any binding of 125I-antithrombin to all regions, including the subendothelium, while leaving the endothelial layer intact.
These data were consistent with the known synthesis of HSPGAT+ by endothelial cells and suggested that the proteoglycans are released from these cells and accumulate predominantly in the basement membrane. This observation suggested two possible ways in which coagulation system activity could be regulated by blood vessel wall HSPGAT+. On the one hand, the small amounts of luminal HSPGAT+ could be in a critical position to bind and activate antithrombin and thereby regulate hemostatic activity at the blood-vessel wall interface. The much larger quantities of abluminal HSPGAT+ could serve as a potential reservoir that could be brought into play with extensive damage to the overlying endothelium. On the other hand, plasma antithrombin may have relatively free access to the much greater amounts of HSPGAT+ that accumulate on the abluminal surface of the endothelial cells, as suggested by studies that document the permeability of the endothelial cell layer.59 Indeed, the presence of 125I-antithrombin bound to subendothelium after ex vivo perfusion suggests that this region is readily accessible to antithrombin. The accessibility of subendothelial HSPGAT+ would also explain kinetic tracer data, which suggest that 10% to 20% of antithrombin partitions to an extravascular compartment.60 These observations imply that coagulation proteases might be inhibited by antithrombin bound to HSPGAT+ in the subendothelium.
Marcum et al.61 obtained evidence that intact blood vessels possess HSPGs that stimulate the activity of plasma antithrombin. In their experiments, rat hindlimb preparations were perfused with purified thrombin at a constant level, and purified antithrombin was infused at several different concentrations. The amount of thrombin-antithrombin complex formed within the vasculature was estimated by a specific radioimmunoassay for the interaction product. The rate of complex formation was enhanced 19-fold in the animal with respect to the uncatalyzed rate in vitro. The antithrombin-stimulating activity detected in the hindlimb vasculature appeared to be due to an HSPG, because the rate of thrombin-antithrombin complex formation was reduced to the uncatalyzed rate by preinfusion of the vasculature with Flavobacterium heparinase. Furthermore, when the hindlimb preparation was perfused with antithrombin chemically modified at Trp49 to decrease its affinity for HS, complex formation also occurred at the uncatalyzed rate. Finally, when purified human platelet factor 4 (PF4) was added to the perfusion stream along with antithrombin, thrombin inhibition occurred at the uncatalyzed rate. These data suggested that PF4, after being released from activated platelets, may play a role in thrombogenesis by neutralizing anticoagulantly active vascular HSPGs. Similar results were obtained with mice genetically deficient in mast cells, strongly suggesting that mast cell heparin plays little or no role in this phenomenon.62
In summary, it appeared likely that a small fraction of plasma antithrombin is normally bound to HSPGAT+ associated with endothelial cells of the blood vessel wall. It was unclear whether the functionally important HSPGs are primarily the small amounts present on the luminal surface of the endothelium, or the much larger quantities located in the subendothelial region, or both. In either case, the endothelial HSPGs could interact with antithrombin to produce an antithrombotic effect at blood-surface interfaces where coagulation proteases are commonly generated. Therefore, antithrombin would be in a position to protect surfaces lined by normal endothelium against thrombus formation. Furthermore, the catalytic nature of anticoagulantly active HS species would ensure the continual regeneration of the nonthrombogenic properties of the endothelial cell layer.
THE PHENOTYPE OF 3-O-SULFOTRANSFERASE-1-DEFICIENT MICE
As discussed in the preceding section, a critical limiting factor in the biosynthesis of HSAT+ is the presence of 3-OST-1, which creates the high-affinity antithrombin-binding motif by modification of selected regions within the HS polymer.54,55 3-OST-5 can also generate HSAT+, although much less efficiently than 3-OST-1.63 Therefore, 3-OST-1 is likely to be the major enzyme responsible for generation of HSAT+ in vivo. Mice lacking the gene that encodes 3-OST-1 (Hs3st1) were generated by HajMohammadi et al.64 to evaluate the biologic role of HSAT+. The absence of 3-OST-1 enzyme caused large reductions (75% to 98%) in HSAT+ levels in most tissues as determined by the ability of HS purified from these tissues to accelerate inhibition of factor Xa by antithrombin. Unexpectedly, removal of most HSAT+ did not alter the basal rate of fibrin generation in 3-OST-1-deficient mice, nor did it lead to enhanced fibrin deposition in the lungs under hypoxemic conditions known to produce a strong procoagulant stimulus. To evaluate the possibility that HSAT+ plays a role only after endothelial injury, which might allow direct contact of the blood with the subendothelial matrix, mice were investigated in a carotid arterial injury model in which application of ferric chloride to the adventitia rapidly induces endothelial denudation leading to thrombosis. However, Hs3st1-/- and Hs3st1+/+ mice had similar thrombotic occlusion times and equivalent postinjury levels of thrombinantithrombin complexes, despite the virtual absence of HSAT+ in the uninjured carotid artery of the Hs3st1-/- mice as determined by 125I-antithrombin binding.
Because 3-OST-1-deficient mice lack an obvious procoagulant phenotype, hemostatic balance does not appear to be tightly correlated with HSAT+ levels. Whereas Hs3st1-/- mice appeared normal on a mixed C57BL/6 × 129S4/SvJae genetic background, intrauterine growth retardation and early postnatal lethality were observed in Hs3st1-/- mice backcrossed into the C57BL/6 background. Although the cause of neonatal mortality in these mice is unclear, histologic examination did not reveal evidence of focal thrombosis as seen in antithrombin-deficient embryos,65 which further suggests a lack of association between 3-OST-1 and antithrombin activity. The unexplained phenotype of Hs3st1-/- mice raises the possibility that HSAT+ participates in a novel biologic function(s). Indeed, recent analyses of Drosophila deficient in the orthologous enzyme, 3-O-sulfotransferase-B, suggest a critical role in Notch signaling pathways.66 Novel functions might even occur outside of the vascular system, because 3-OST-1-derived HSAT+ is produced in several nonendothelial cell types, including
fibroblastic cell lines,67 ovarian granulosa cells,68 embryonal carcinoma cells,57 alveolar type II cells,69 and glomerular epithelial cells.70 The lack of a prothrombotic phenotype in 3-OST-1 knockout mice challenges the concept that HSAT+ plays a major role in hemostasis. However, unprovoked thrombosis occurs in humans or mice homozygous for antithrombin mutations that reduce the inhibitor’s affinity for HS.71,72 These findings suggest that HS binding is required for proper antithrombin function in vivo. How, then, could one explain the 3-OST-1 knockout data?
fibroblastic cell lines,67 ovarian granulosa cells,68 embryonal carcinoma cells,57 alveolar type II cells,69 and glomerular epithelial cells.70 The lack of a prothrombotic phenotype in 3-OST-1 knockout mice challenges the concept that HSAT+ plays a major role in hemostasis. However, unprovoked thrombosis occurs in humans or mice homozygous for antithrombin mutations that reduce the inhibitor’s affinity for HS.71,72 These findings suggest that HS binding is required for proper antithrombin function in vivo. How, then, could one explain the 3-OST-1 knockout data?
First, the residual 2% to 20% anticoagulant HS activity in the 3-OST-1-deficient mice likely reflects 3-O-sulfation by 3-O-sulfotransferase isoforms other than 3-OST-1. As indicated in the preceding text, 3-OST-5 might play an important role if hemostatic balance requires only low levels of HSAT+.63 Because the plasma antithrombin concentration (2 to 5 µM) greatly exceeds the dissociation constant of antithrombin for HSAT+ (˜15 nM), small amounts of HSAT+ might always be saturated with antithrombin and thereby exert a basal anticoagulant tone. Assessment of this possibility will require deletion of the gene for 3-OST-5 and perhaps other isoforms that may be responsible for the synthesis of the HSAT+ present in Hs3st-/- mice.
Second, HSAT+ may not be responsible for the anticoagulant properties of HS in vivo. Scully et al.73 reported that HS that failed to bind to a column of immobilized antithrombin (generally defined as HSAT-) accelerated the factor Xa-antithrombin reaction by 1,100-fold, although much higher concentrations were required in comparison with high-affinity HS (HSAT+). They proposed that the nonthrombogenic property of blood vessels is due to acceleration of the factor Xa-antithrombin interaction by the greater mass of vascular HSAT- rather than by the much smaller proportion of HSAT+ molecules. If this hypothesis is true, one would predict that defective HS synthesis would lead to a procoagulant phenotype. To complicate matters, HS interacts with a great variety of proteins other than antithrombin, and multiple functions have been attributed to HS throughout embryonic development, including interactions with the FGF, Wnt, transforming growth factors-β (TGF-β), and hedgehog (Hh) signaling pathways.74 Targeted disruption of EXT1, one of the genes responsible for HS chain polymerization, resulted in the loss of HS, disruption of gastrulation, and embryonic lethality before embryonic day 8.5.75 Conditional knockouts of EXT1 or other HS biosynthetic genes in vascular tissue at later stages of development could be more informative with regard to the hemostatic properties of HS but have not yet been achieved. Knockout of the syndecan-4 gene (Synd4) suggests that HS might be involved in anticoagulation of the placenta.76 Syndecan-4, a transmembrane protein bearing both HSAT+ and HSAT- chains, was detected in the fetal vessels of the placenta by in situ hybridization and immunohistochemical staining. During late gestation, degeneration of fetal vessels in the placental labyrinth was more extensive in Synd4-/- embryos than in wild-type controls and was associated with deposition of fibrin(ogen). These findings suggest that the absence of syndecan-4 produces a deficit in placental anticoagulation.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


