INTRODUCTION
SUMMARY
Hemophilias A and B are the only two bleeding disorders inherited in a sex-linked fashion. The gene for both disorders is on the long arm of the X-chromosome. Both disorders appear as otherwise clinically indistinguishable hemorrhagic diseases of mild, moderate, or life-threatening severity. In the most-severe form, both hemophilias A and B are characterized by multiple bleeding episodes into joints and other tissues leading to chronic crippling hemarthropathy and internal organ hemorrhage unless treated early or prophylactically with factor VIII or IX concentrates, respectively. Even though phenotypically similar, both diseases are genetically heterogeneous with more than 1000 mutations leading to the absence of or dysfunctional factor VIII or IX molecules that do not support normal thrombin generation nor adequate fibrin clot formation.
Despite similarities in hemorrhagic symptoms, there are major differences between hemophilias A and B. Hemophilia A is about five times more common than hemophilia B, and is caused by defects in the factor VIII gene, a large 186-kb gene with 26 exons. A common mutation results from inversion and crossing over of intron 22 during meiosis. This mutation leads to severe hemophilia, and because no factor VIII protein is made, these patients are prone to developing antibody inhibitors to therapeutically administered factor VIII that neutralize its coagulant function, making adequate therapy problematic. Approximately 20 percent of severely affected hemophilia A patients develop such inhibitors, whereas only 3 percent or fewer of severely affected hemophilia B patients develop inhibitors against factor IX. About one-third of the mutations in hemophilias A and B arise de novo at CpG “hotspots.” These mutations are apt to occur in the germ cells of a maternal grandfather whose daughters will be carriers and whose grandsons will have a 50 percent chance of having hemophilia.
Replacement therapy is available for both hemophilia A and hemophilia B patients. Safe, effective, and highly purified factor VIII and factor IX concentrates derived from plasma or made by recombinant technology are available for prophylactic therapy to prevent bleeding episodes or prompt treatment of hemorrhagic events. Prophylaxis is the treatment of choice and can prevent disabling joint disease and other hemorrhagic events such that patients can expect a relatively normal life span provided that adequate replacement therapy is available. For patients with inhibitors, factor VIIa and factor VIII inhibitor bypassing activity can be used to “bypass” the factor VIII or factor IX deficiency. Both disorders are good candidates for gene therapy that may eventually lead to their cure.
Acronyms and Abbreviations
AAV, adeno-associated virus; aPTT, activated partial thromboplastin time; BT, bleeding time; BU, Bethesda unit; CGA, cytosine, guanine, adenine; CJD, Creutzfeldt-Jakob disease; COX, cyclooxygenase; CRM, cross-reacting material; CT, computerized tomography; DDAVP, 1-desamino-8-d-arginine vasopressin, desmopressin; DVT, deep vein thrombosis; EACA, ε-aminocaproic acid; FEIBA, factor VIII inhibitor bypassing activity; GLA, γ-carboxyglutamic acid; Ig, immunoglobulin; PT, prothrombin time; PTC, plasma thromboplastin component (factor IX); RFLP, restriction fragment length polymorphism; TCT, thrombin clotting time; VWD, von Willebrand disease; VWF, von Willebrand factor.
HEMOPHILIA A (CLASSIC HEMOPHILIA, FACTOR VIII DEFICIENCY)
Hemophilia A is an X-linked hereditary disorder caused by defective synthesis of factor VIII. Hemophilia A is less common than von Willebrand disease (VWD; Chap. 126), but it is more common than other inherited clotting factor abnormalities. The estimated incidence of hemophilia A is one in every 5000 to 7000 live male births. It occurs in all ethnic groups in all parts of the world.1
Sex-linked hemophilia was recognized at least as early as the 2nd century, when a rabbi correctly deduced that sons of hemophilic carriers were at risk for bleeding following circumcision.2 In the 19th century, several authors noted the sex-linked inheritance pattern of the disease and ascribed the hemorrhagic episodes to delayed blood coagulation. Morawitz3 developed the classic theory of blood coagulation, which recognized two major reactions: (1) conversion of prothrombin to thrombin by a tissue substance that Morawitz termed thrombokinase, and (2) conversion of fibrinogen to fibrin by thrombin. In 1911, Addis4 demonstrated that thrombin formed more slowly in hemophilic blood than in normal blood and that the defect could be corrected by small amounts of normal plasma. However, he incorrectly theorized that hemophilia resulted from prothrombin deficiency. As protein purification techniques improved throughout the 1930s and 1940s, thrombokinase was resolved into several distinct components. Brinkhous5 demonstrated that the prothrombin content of hemophilic plasma was normal and that the basic defect in hemophilia was the delayed conversion of prothrombin to thrombin. The defect could be corrected by a fraction of normal plasma containing the antihemophilic factor, later named factor VIII. In 1947, Pavlovsky6 observed that when blood from one patient with hemophilia was transfused into another patient with a similar clinical phenotype, the prolonged clotting time in the recipient was corrected. At the time, Pavlovsky did not recognize that he was dealing with two different types of hemophilia. This fact was recognized by Aggeler and coworkers7 in 1952, when they described a patient deficient in “plasma thromboplastin component”, a blood clotting factor different from factor VIII. A deficiency of “plasma thromboplastin component,” later termed factor IX, was identified as the cause of hemophilia B. A month later, Biggs and colleagues described a similar patient whose surname was Christmas, thus the synonym “Christmas disease.”8 Hemophilias A and B are the only two hereditary clotting factor defects inherited in a sex-linked pattern, and they are clinically indistinguishable, although data suggest that on the whole, hemophilia B may be less severe than hemophilia A.9 However, in an individual patient, the disorders cannot be distinguished without a specific assay for factor VIII or IX.
In 1964, a proposal to organize the growing number of coagulation factors into a cascade or waterfall mechanism was put forth by Davie and Ratnoff and by Macfarlane.10,11 In this scheme, each zymogen clotting factor was sequentially activated to a protease that subsequently activated the next zymogen until thrombin ultimately was produced. In this scheme, factors VIII and IX were considered to be proenzymes. Later, however, factor VIII, when activated by thrombin, was shown not to be a proenzyme but rather an essential cofactor for factor IXa. The waterfall hypothesis has been modified so that the primary role of the tissue factor–factor VII complex in the initiation of coagulation is emphasized (Chap. 113).12
Hemophilia A is a heterogeneous disorder resulting from defects in the factor VIII gene that leads to absent or reduced circulating levels of functional factor VIII. The reduced activity can result from a decreased amount of factor VIII protein, the presence of a functionally abnormal protein, or a combination of both. For factor VIII to be an effective cofactor for factor IXa, it must first be activated by thrombin, a reaction that results in the formation of a heterotrimer composed of the A1, A2, A3, C1, and C2 domains of factor VIII in a complex with calcium (Chap. 113).13 Activated factor VIII (factor VIIIa) and activated factor IX (factor IXa) associate on the surface of activated platelets, forming a functional factor X-activating complex (“tenase” or “Xase”).14 In the presence of factor VIIIa, the rate of factor X activation by factor IXa is dramatically enhanced. That hemophilia A and hemophilia B have similar clinical manifestations is not surprising, because both factor VIIIa and factor IXa are required to form the Xase complex. The lack of either activated protein leads to a similar lack of platelet surface Xase activity with subsequent decreased thrombin generation. In patients with hemophilia, clot formation is delayed because of the decreased thrombin generation. The clot that is formed is friable, easily dislodged, and highly susceptible to fibrinolysis, all of which lead to excessive bleeding and poor wound healing.15
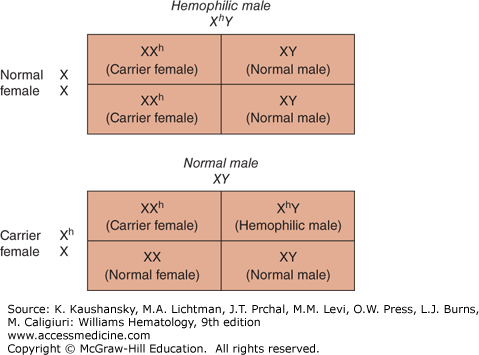
Hemophilia A results when mutations occur in the factor VIII gene located on the long arm of the X-chromosome (X-q28). The disease occurs almost exclusively in males. Figure 123–1 shows the inheritance pattern of hemophilia A and hemophilia B. All the sons of affected hemophilic males are normal, whereas all the daughters are obligatory carriers of the factor VIII defect. Sons of carriers have a 50 percent chance of being affected, whereas daughters of carriers have a 50 percent chance of being carriers themselves.
Figure 123–1.
Inheritance pattern of hemophilia. All daughters of a hemophilic male are carriers of hemophilia, whereas all sons are normal. Daughters of carriers have a 50 percent chance of being a carrier, whereas sons of carriers have a 50 percent chance of having hemophilia. X, normal; Xh, abnormal X chromosome with the hemophilic gene; XhY, hemophilic male; XX, normal female; XXh, carrier female; XY, normal male; Y, normal.
The factor VIII gene is very large, approximately 186 kb, with approximately 9 kb of exons. The gene contains 26 exons and 25 introns.16 Based on the sequence of the factor VIII gene in normal individuals and patients with hemophilia A, numerous specific mutations have been described16,17; as of 2015, more than 2000 specific variants in the factor VIII gene resulting in classic hemophilia have been described.17
Hemophilia A can result from multiple alterations in the factor VIII gene. These include gene rearrangements; missense mutations, in which a single base substitution leads to an amino acid change in the molecule; nonsense mutations, which result in a stop codon; abnormal splicing of the gene; deletions of all or portions of the gene; and insertions of genetic elements.18 The genetic defects leading to hemophilia have been reviewed.17
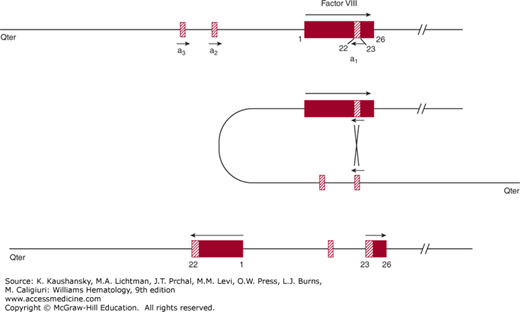
One of the most common mutations, accounting for 40 to 50 percent of severe hemophilia A patients, is a unique “combined gene inversion and crossing over” that disrupts the factor VIII gene.19,20 Figures 123–2 and 123–3 schematically depict the factor VIII gene and the mechanism of the “inversion–crossing over.”21 Within intron 22 are two other genes: (1) F8A(a1), which is transcribed in the 5′ direction, and (2) F8B, which is transcribed in the 3′ direction of the factor VIII gene. The hatched boxes in Figure 123–3 show two other extragenic homologous sequences (a2,a3) 5′ to the F8A gene that lies within intron 22 (a1). The presence of extragenic F8A sequences 5′ to the F8A gene within intron 22 is central to the inversion and translocation of part of the factor VIII gene from exon 1 to exon 22. The mechanism is homologous recombination between the F8A sequence that lies within intron 22 and one of the homologous extragenic sequences of the F8A gene 5′ to the factor VIII gene. During meiosis, crossing over of homologous sequences occurs between the F8A gene lying within intron 22 and one of the extragenic homologous F8A sequences 5′ to intron 22. Thus, the transcription of the complete factor VIII sequence is interrupted (Fig. 123–3). Figure 123–3 shows a common inversion and crossing over, but homologous recombinations can occur with either of the extragenic genes. Approximately 2 to 5 percent of the severe cases of hemophilia A carry the intron 1 inversion resulting in the separation of the F8 promoter-exon 1 sequence from the remainder of the F8 gene.22 The “inversion–crossing over” mutations result in severe hemophilia, and approximately 20 percent of these patients are susceptible to developing antibody inhibitors that neutralize factor VIII coagulant function.
Figure 123–2.
Schematic of the factor VIII gene (FVIII). The FVIII gene is located at q28 on the long arm of the X chromosome. The region of the FVIII gene is enlarged on the second line. Note that two genes, designated a2 and a3, are 5′ to the FVIII gene. The hatched area indicated on FVIII corresponds to intron 22 shown on the third line. Within intron 22 (fourth line) are two nested genes, one designated F8A, which is transcribed in a direction opposite to that of the whole FVIII and is homologous to the a2 and a3 genes shown on line 2. G6PD, glucose-6-phosphate dehydrogenase. (Reproduced with permission from Scriver CR, Beaudet AL, Sly WS et al: Metabolic and Molecular Basis of Inherited Diseases, 8th ed. McGraw-Hill, New York, 1995.)
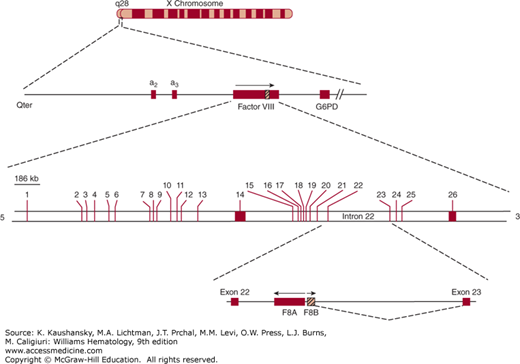
Figure 123–3.
Schematic of inversion and crossing over at intron 22. Inversion and crossing-over of the a3 gene with its homologous sequence a1 nested within intron 22 are shown. Middle panel: When crossing over of the a1 gene nested within intron 22 and the a3 gene extragenic to FVIII occurs, a portion of FVIII is transcribed in a reverse manner from exon 1 through exon 22. Homologous recombination with the extragenic a2 gene is also possible. In some individuals there are two a2 or a3 extragenic sequences giving rise to four possible types of the “inversion–crossing over” mechanism. (Reproduced with permission from Antonarakis SE, Kazazian HH, Tuddenham EG: Molecular etiology of factor VIII deficiency in hemophilia A. Hum Mutat 1995;5(1):1–22.)
Of the different insertions in the factor VIII gene that have been reported, a few are long interspersed elements (LINEs) that are transposon sequences; that is, sequences that have been inserted frequently throughout the genome.23 Most of these insertions result in severe hemophilia.
In many cases of hemophilia, there is no family history of the disease, and at least 30 percent of the cases of hemophilia are a result of spontaneous (de novo) mutations. Most of these occur at CpG dinucleotides in the factor VIII gene.23 De novo occurrences of hemophilia usually result from a mutation in the gamete of a normal male; for example, a mutation in the germ cell of a maternal grandfather will give rise to the hemophilia gene in his daughters such that his grandsons may have hemophilia.18 Codons for the amino acid arginine (CGA [cytosine, guanine, adenine]) are frequently affected by mutations at CG doublets. A C→T transition often results in a stop codon with synthesis of a truncated factor VIII molecule and usually is associated with severe hemophilia A. However, a G→A transition results in a missense mutation, which often leads to a dysfunctional factor VIII molecule that may be associated with mild, moderate, or severe hemophilia. Some missense mutations result in the production of normal or near-normal amounts of factor VIII antigen, while the coagulant activity may be dramatically or only slightly reduced. Many other single-base substitutions have been described, resulting in hemophilia of varying degrees of severity.
Large deletions in the factor VIII gene almost always are associated with severe hemophilia. On the other hand, a small deletion that does not change the reading frame of the gene may result in milder disease. Patients with large deletions who have no detectable factor VIII antigen are more susceptible to the development of anti–factor VIII antibodies, although antibodies clearly also occur in patients without deletions.16,23
Hemophilia A in females is extremely rare, although an affected female offspring from a hemophilic father and carrier mother have been reported. Hemophilia A may occur in females with X chromosomal abnormalities such as Turner syndrome, X chromosomal mosaicism, and other X chromosomal defects.23,24 If the normal X chromosome is inactivated disproportionately (“imbalanced X inactivation”) in a carrier female, factor VIII levels may be sufficiently low to cause bleeding manifestations. Usually these manifestations are mild, but they may be serious during surgical procedures or following significant trauma.
A careful and complete family history is important for carrier detection.25 All daughters of a hemophilic father are obligatory carriers of the hemophilic defect. If a known carrier has a daughter, that daughter has a 50 percent chance of being a carrier.
Carrier detection is important when a daughter of a known carrier or a female offspring of a hemophilic patient wishes to become pregnant. At times, the history of hemophilia in the family is in a distant blood relative, and the gene for hemophilia may skip several generations. The current standard for identifying carrier status is through direct gene sequencing. Carriers who harbor the intron 22 inversion or intron 1 inversion can be identified using the Southern blot technique and polymerase chain reaction, respectively.22,25 If these mutations are found to be absent, sequencing of the complete coding region is performed.26
Use of markers for restriction fragment length polymorphism (RFLP) is simpler than direct sequencing of the coding region of the factor VIII gene, but use of the RFLP technique requires that the pedigree analyses include at least one hemophilic male whose mother is heterozygous for one or more RFLP markers.27,28 This technique is no longer considered to be the optimal approach in genotyping of affected males or carrier females.
Prenatal diagnosis of hemophilia now can be performed almost routinely.29 If a carrier female has a fetus that can be identified as a female by chromosomal analysis of cells obtained by amniocentesis (at approximately 16 weeks of gestation), analysis of free fetal DNA, ultrasound or by chorionic villus sampling at week 10 of gestation, little concern exists regarding whether the female fetus is a carrier because carriers usually have no bleeding tendency. If the fetus is a male, sufficient cells can be obtained to perform DNA analysis using the methods described above. The decision on whether to carry an affected male fetus to term should be decided by the parents after they are appropriately counseled and provided with all the necessary genetic, clinical, and therapeutic information about hemophilia. As the treatment for hemophilia A improves, the decision to continue an affected pregnancy should become far easier.
Hemophilia A is characterized by excessive bleeding into various tissues of the body, including soft-tissue hematomas and hemarthroses that can lead to severe crippling hemarthropathy. Recurrent hemarthroses are characteristic of the disease. The disease has been broadly classified as mild, moderate, and severe, although overlap exists between these categories. Table 123–1 shows a classification based on the severity of clinical manifestations. A range of plasma factor VIII concentrations in percentages of normal and in units per milliliter is given for each category. Approximately 10 percent of individuals with factor VIII levels compatible with severe hemophilia may exhibit milder symptoms.30 Among other explanations, this phenotypic heterogeneity could be a result of coinheritance of thrombophilic mutations, such as the factor V Leiden mutation (R506Q).31 Severely affected patients (<1 percent factor VIII) frequently experience “spontaneous” bleeding without known trauma other than that associated with the usual day-to-day activities. Without effective treatment, recurrent hemarthroses, resulting in chronic hemophilic arthropathy, occur by young adulthood and are highly characteristic of the severe form of the disorder. However, bleeding episodes are intermittent, and some patients do not bleed for weeks or months. Except for intracranial bleeding, sudden death because of hemorrhage is rare in societies where clotting factor concentrates are freely available.
| Classification | Factor VIII Level | Clinical Features |
|---|---|---|
| Severe | ≤1% of normal (≤0.01 U/mL) | 1. Spontaneous hemorrhage from early infancy |
| 2. Frequent spontaneous hemarthroses and other hemorrhages, requiring clotting factor replacement | ||
| Moderate | 1–5% of normal (0.01–0.05 U/mL) | 1. Hemorrhage secondary to trauma or surgery |
| 2. Occasional spontaneous hemarthroses | ||
| Mild | 6–40% of normal (0.06–0.40 U/mL) | 1. Hemorrhage secondary to trauma or surgery |
| 2. Rare spontaneous hemorrhage |
Moderately affected patients with hemophilia may have occasional hematomas. Hemarthroses, usually associated with a known trauma, may occur as well. These patients have greater than 1 percent but less than 5 percent of normal factor VIII activity.
Mildly affected patients with hemophilia, who have factor VIII levels between 6 to 40 percent, have infrequent bleeding episodes. The disease may go undiagnosed and be discovered only because of excessive hemorrhage postoperatively, following trauma, or after the toss and tumble of contact sports.
Most carriers have approximately 50 percent factor VIII activity and experience no bleeding symptoms, even with surgical procedures. Carriers with factor VIII levels significantly less than 50 percent, as a result of imbalanced X chromosome inactivation, may experience excessive bleeding after trauma (e.g., childbirth or surgery). Therefore, measurement of factor VIII level is recommended in all carriers.
Bleeding into joints accounts for approximately 75 percent of bleeding episodes in severely affected patients with hemophilia A.32,33 The normal synovium has few cells, but numerous capillaries beneath the synovial layer can be damaged by the mechanical trauma associated with daily use of joints. The joints most frequently involved, in decreasing order of frequency are knees, elbows, ankles, shoulders, wrists, and hips. Hinge joints are much more likely to be involved than are ball-and-socket joints. Hemarthroses usually occur when an affected child begins to walk.
Hemarthroses are heralded by an aura of mild discomfort that, over a period of minutes to hours, becomes progressively painful. The joint usually swells, becomes warm, and exhibits limited motion. Occasionally, the patient experiences a mild fever. Significant and sustained fever, however, suggests an infected joint. When joint bleeding does not respond to replacement therapy, one should suspect the presence of an inhibitor of factor VIII or an infected joint. Bleeding into the knee joint is more easily detected by physical findings than is bleeding into either the elbow or shoulder. When bleeding stops, the blood resorbs, and the symptoms gradually subside over a period of several days. If hemarthroses are treated early, pain usually subsides in 6 to 8 hours and disappears in 12 to 24 hours. However, repeated hemorrhage into the joints eventually results in extensive destruction of articular cartilage, synovial hyperplasia, and other reactive changes in the adjacent bone and tissues. Iron deposits from residual blood is a major factor in the pathogenesis of hemophilic arthropathy.33 Acute bleeding into a chronically affected joint may be difficult to distinguish from the pain of degenerative arthritis.
A major complication of repeated hemarthroses is joint deformity complicated by muscle atrophy and soft-tissue contractures (Fig. 123–4). Figure 123–5 shows the various radiologic stages of progressive destruction of joint cartilage and adjacent bone. Osteoporosis and cystic areas in the subchondral bone may develop, and progressive loss of joint space occurs. Figure 123–6 shows a magnetic resonance image (MRI) of a normal knee in comparison to a knee from an individual with severe hemophilia with arthropathy. Figure 123–7 depicts bleeding into a hemophilic ankle.
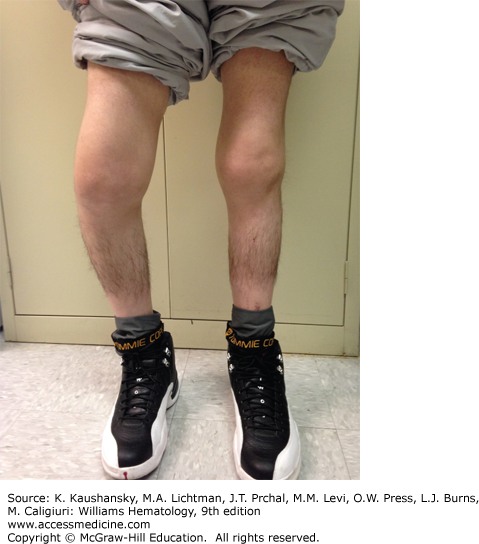
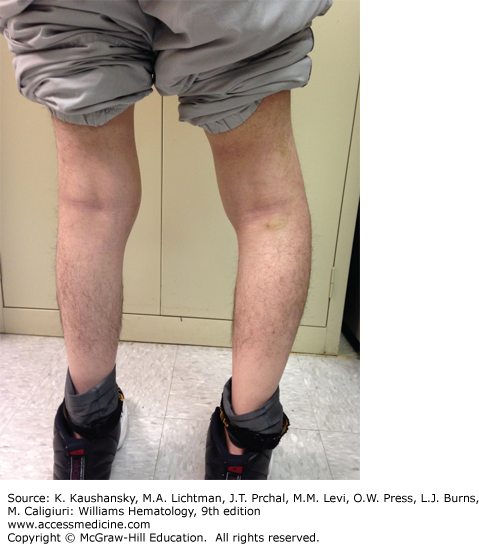
Figure 123–5.
Various radiologic stages of hemophilic arthropathy. Stages 0 (normal joint) and 1 (fluid in the joint) are not shown. A. Stage 2. Some osteoporosis and epiphyseal overgrowth are present in knee 2. Epiphysis is wider in knee 2 than in knee 1 (arrows). B. Stage 3. Subchondral bone cysts (arrowheads). Joint spaces exhibit irregularities. C. Stage 4. Prominent bone cysts with marked narrowing of joint space (arrow). D. Stage 5. Obliteration of joint space with epiphyseal overgrowth (arrow).
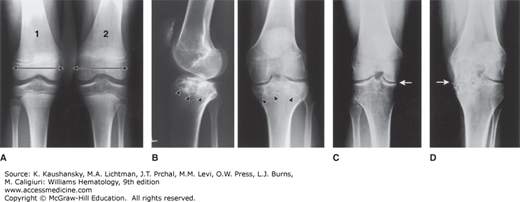
Figure 123–6.
Magnetic resonance imaging (MRI) of normal and hemophilic knees. A. MRI of normal knee. B. A transverse T2-weighted spin-echo image of the knee shows an effusion (*) and multiple foci of hemosiderin deposition (arrows) along the synovium lining the suprapatellar bursa. C. A sagittal T2-weighted spin-echo image of the knee shows dark foci of synovial hemosiderin deposition (white arrows) accompanied by narrowing of the femorotibial joint (black arrow). D. A sagittal STIR (short tau inversion recovery) image of the knee (in the same patient as B) demonstrates an effusion in the suprapatellar bursa (asterisks). The irregular, lumpy surface of the bursa represents thickened, hemosiderin-laden synovium. Femorotibial joint narrowing (black arrow) is associated with edema in the subchondral bone of the femoral condyle (white arrow). (Used with permission of Dr. Jordan Renner, University of North Carolina.)
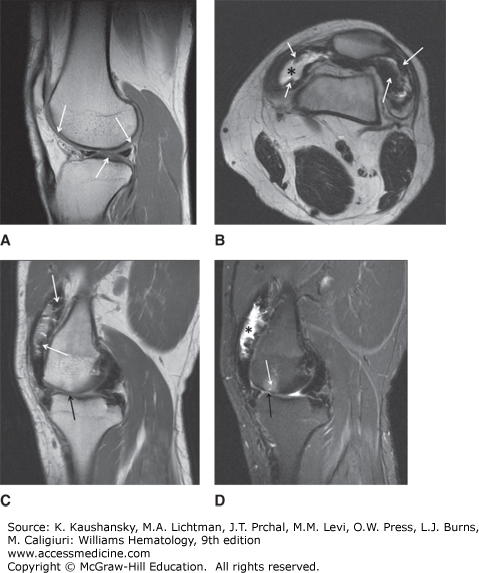
Figure 123–7.
A. A sagittal STIR (short tau inversion recovery) image of an ankle shows an effusion (white arrow). Edema in the distal tibia (asterisks) surrounds a debris-filled defect in the subchondral bone of the distal tibia (black arrows). B. A coronal proton density of the ankle in the same patient as in A shows the defect in the subchondral bone of the distal tibia (white arrow). Mild narrowing of the tibiotalar joint (black arrows) is more apparent laterally.
Repeated bleeding into a joint results in synovial hypertrophy and inflammation. The synovium is thickened and folded, leading to limited joint motion. The result is a tendency for repeated hemorrhages leading to a so-called target joint.32 Indeed, a target joint is defined by the occurrence of three or more spontaneous bleeds within a 6-month period. The joints most often involved are the knees, ankles, and elbows, which become chronically swollen. Chronic synovitis may persist for months or years unless the condition is adequately treated.
Infection of hemophilic joints is not common but must be suspected in all patients with fever, leukocytosis, or other systemic manifestations. Rapid diagnosis is mandatory, because infection of such joints leads to rapid loss of joint architecture and function. A painful and swollen joint may require aspiration, which should be performed by experienced personnel using meticulous aseptic techniques and appropriate factor replacement therapy prior to aspiration.
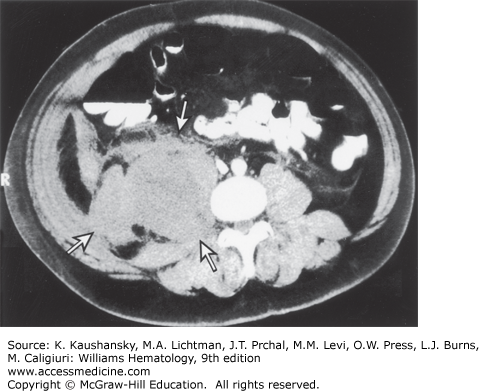
Soft-tissue hematomas are also characteristic of hemophilia A. Hemorrhage into subcutaneous connective tissues or into muscles may occur with or without a known trauma. Hematomas, once formed, may stabilize and slowly resorb. However, in moderately and severely affected patients, hematomas have a tendency to enlarge progressively and to dissect in all directions, unless appropriately treated. Rarely, retroperitoneal hematomas, after beginning in the iliopsoas muscle, can dissect superiorly through the diaphragm, into the chest, and sometimes even into the soft tissues of the neck, compromising the airway. A retroperitoneal hematoma is more likely to compromise renal function by causing ureteral obstruction. Figure 123–8 shows the computed tomography (CT) scan of a patient with a retroperitoneal hemorrhage. Other hematomas expand locally and may compress adjacent organs, blood vessels, and nerves. A rare, and often fatal, complication of an abdominal hematoma is perforation and drainage into the colon. Subcutaneous hematomas may dissect into muscle. Pharyngeal and retropharyngeal hematomas, sometimes complicating simple colds, may enlarge and obstruct the airway. Hemorrhage in or around the airway is a potentially life-threatening situation that requires prompt administration of factor VIII.
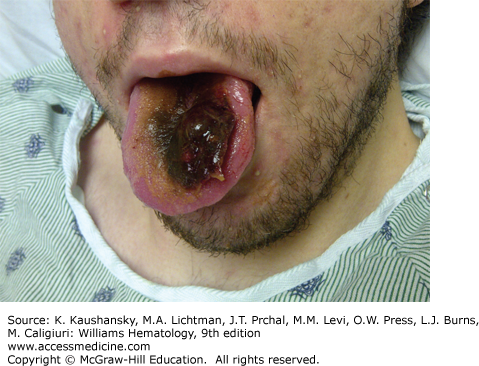
Hemorrhages occur into muscle in the following order of frequency: calf, thigh, buttocks, and forearm. Recurrent or unresolved hematomas may lead to muscle contractures, nerve palsies, and muscle atrophy. Bleeding into the tongue (Fig. 123–9) or frenulum is particularly frequent in young children and usually is caused by trauma.
Bleeding into fascia and muscle can result in a so-called compartment syndrome. This results when hemorrhage in a confined space compresses the arterial vasculature resulting in ischemic muscle injury. Compartment syndrome tends to occur in the distal part of the extremities, particularly in the flexor muscles, and sometimes requires urgent fasciotomy under cover of clotting factor replacement therapy. Bleeding into the myocardium or erect penis is very unusual, perhaps explained by the high concentration of tissue factor in these tissues.
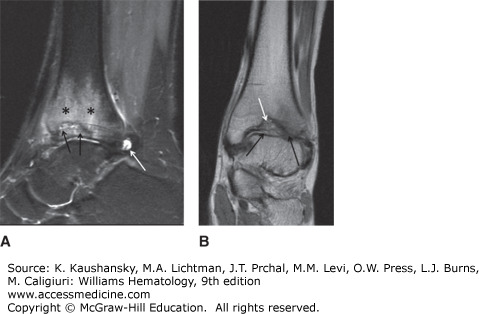
Pseudotumors are blood cysts that occur in soft tissues or bone. They are rare but dangerous complications of hemophilia (Fig. 123–10).34 They are classified into three types. One type is a simple cyst that is confined by tendinous attachments within the fascial envelope of a muscle. The second type initially develops as a simple cyst in soft tissues such as a tendon, but it interferes with the vascular supply to the adjacent bone and periosteum, resulting in cyst formation and resorption of bone. The third type is thought to result from subperiosteal bleeding that separates the periosteum from the bony cortex. Most pseudotumors are not associated with pain unless rapid growth or nerve compression occurs. As the volume of the cyst increases, the cyst compresses and destroys the adjacent muscle, nerve, and/or bone or expands around structures like ureters causing renal failure. Pseudotumors usually contain either serosanguineous fluid or a viscous brownish material surrounded by a fibrous membrane (Fig. 123–10). Pseudotumors have a tendency to expand over several years and eventually become multiloculated. Some reach enormous size and involve so many structures that make them inoperable. Erosion through surrounding tissues and penetration into viscera or through the skin can occur, usually as a late event. Sinus tracts from the pseudotumor predispose to infection and septicemia. Pseudotumors often develop in the lower half of the body, usually in the thigh, buttock, or pelvis, but they can occur anywhere, including the temporal bone. CT or MRI is useful for diagnosis. Needle biopsies of pseudotumors should be avoided because of the risk of infection and hemorrhage. A reliable treatment is operative removal of the entire mass because the pseudotumor likely will reform if it is not completely removed. Embolization, percutaneous drainage, and radiotherapy of a pseudotumor have been reported and may be of value in hemophiliacs with inhibitors when surgery is not possible.35 Surgical treatment of patients with large pseudotumors should be done in a hemophilia treatment center with a specialized multidisciplinary team of experts.36
Figure 123–10.
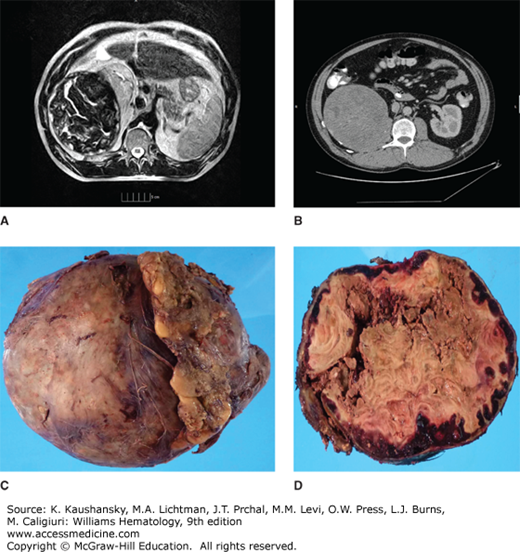
Retroperitoneal pseudotumor. A and B. Magnetic resonance imaging and computed tomography scan of pseudotumor arising from the iliopsoas muscle compressing the kidney and other adjacent structures. Loculations and calcifications can be seen. C. Gross specimen after surgical removal, weighting approximately 6 pounds. D. Cross-section of pseudotumor shows peripheral red hemorrhage, centrally caseified blood and necrosis. Note the thick capsule that surrounds the tumor.
Many severely affected patients with hemophilia experience episodes of spontaneous and asymptomatic hematuria. The urine may be brown or red, depending upon the rate of bleeding. Most bleeding arises from the renal pelvis, usually from one kidney but occasionally from both. Appropriate studies to exclude a structural lesion in the kidneys should be performed. Administration of factor replacement and hydration is usually sufficient to arrest the bleeding. Antifibrinolytic agents, such as aminocaproic acid and tranexamic acid should be avoided in individuals with hematuria because of the risk of forming clots and producing obstructing clots in the ureter.
Intracranial bleeding is one of the most dangerous hemorrhagic events in hemophilic patients.37 Currently, bleeding into the brain is a leading cause of death in hemophilic patients. Hemorrhage into the central nervous system may be “spontaneous” but usually follows trauma, which may be trivial. Symptoms often occur soon after trauma, but sometimes are delayed. For example, symptoms of a subdural hematoma may be delayed for days or several weeks. Hemorrhage into the brain parenchyma or a subdural or epidural hematoma should always be suspected in hemophilic patients with unusual headaches. When intracranial bleeding is suspected, the patient should be treated immediately with factor VIII and diagnostic procedures, such as CT scans or MRI studies should be delayed until after treatment is initiated. Although lumbar puncture has been performed safely in severe hemophilic patients without replacement therapy, replacing factor VIII to a level of approximately 50 percent of normal prior to the procedure is advisable.
Hemorrhage into the spinal canal is an uncommon neurologic complication in hemophilia, mostly related to trauma that can result in paraplegia. Bleeding may occur within the spinal cord itself, but epidural bleeding compressing the cord is more common.
Peripheral nerve compression is a frequent complication of muscle hematomas, particularly in the extremities. Compression of the femoral nerve by a hematoma in the iliopsoas muscle can result in sensory loss over the lateral and anterior thigh, weakness and atrophy of the quadriceps, and loss of the patellar reflex. The ulnar nerve is the next most frequently involved peripheral nerve. Bleeding may occur in any muscle and may compress local neural blood supply. This situation can be followed by permanent neuromuscular defects and multiple contractures.
Mucous membrane bleeding is common in hemophilia. Epistaxis and hemoptysis, often resulting from allergic reactions or trauma, can be associated with local structural lesions involving the upper and/or lower respiratory tract. Treatment of epistaxis by cautery or nasal packing sometimes is followed by recurrent bleeding because of sloughing of the cauterized area or dislodging of a poorly formed clot when the packing is removed. Gastrointestinal hemorrhage has a 1.3 percent annual incidence and is mostly associated to older age and complications of advanced liver disease. Ingestion of antiinflammatory drugs for relief of pain of hemophilic arthropathy is a frequent cause of upper gastrointestinal hemorrhage, and a history of ingestion of aspirin and other antiinflammatory drugs should be specifically addressed (and proscribed) when assessing the etiology of such bleeding.38
Hemophilic patients are treated with clotting factor preoperatively and postoperatively to prevent bleeding. Mildly or sometimes moderately affected patients may go unrecognized until surgery results in excessive bleeding at the surgical site. Bleeding may be delayed for several hours or, occasionally, for several days. Surgery in such patients is characterized by delayed wound healing because of poor clot formation.15 Prolonged bleeding and subsequent infection of the wound hematoma may further complicate healing. Appropriate factor VIII replacement therapy, sometimes supplemented by anti-fibrinolytic agents, can prevent intraoperative and postoperative hemorrhages.
Dental extraction is the most frequent surgical procedure performed on hemophilic patients. Loss of deciduous teeth seldom causes excessive bleeding, but extraction of permanent teeth may result in excessive hemorrhage that can persist intermittently for several days to weeks unless appropriate treatment is administered. In the untreated patient with severe hemophilia, life-threatening, dissecting pharyngeal and/or sublingual hematomas may result from dental procedures or from administration of regional block anesthesia.
Patients with severe hemophilia A have a prolonged activated partial thromboplastin time (aPTT). The prothrombin time (PT) and thrombin clotting time (TCT) are normal. Different combinations of aPTT reagents and instrumentation exhibit varying sensitivities to factor VIII levels. In mild hemophilia, the aPTT may be only slightly prolonged or at the upper limit of normal, especially if factor VIII activity is 20 percent or greater. The aPTT is corrected when hemophilic plasma is mixed with an equal volume of normal plasma. If the hemophilic plasma contains an anti-factor VIII inhibitor antibody, the aPTT on a similar mixture is prolonged, but incubation of the mixture for 1 or 2 hours at 37°C is sometimes required to detect the prolongation. A definitive diagnosis of hemophilia A should be based on a specific assay for factor VIII activity.
Functional factor VIII coagulant activity is measured by one-stage clotting assays based on the aPTT. Chromogenic assays for factor VIII activity also are used widely, but do not always agree with one-stage assays.39 Although infrequently measured in practice, factor VIII antigen is measured by immunologic assays, which detect normal and most abnormal factor VIII molecules. If the factor VIII antigen level is normal but the clotting activity is reduced, the patient has a dysfunctional factor VIII molecule. Such patients have antigen-positive hemophilia, also referred to as cross-reacting material (CRM)-positive.40 Patients in whom both the factor VIII antigen level and activity are nearly undetectable are said to be CRM-negative.
VWD sometimes is confused with hemophilia A. The basic defect in VWD is reduced activity of von Willebrand factor (VWF), which acts as a carrier of factor VIII in vivo (Chap. 126). Thus, in VWD, factor VIII levels are reduced, although considerable variability exists. Although factor VIII is synthesized normally in patients with VWD, the half-life of factor VIII is markedly shortened because the VWF “carrier” molecule is decreased or absent. Other abnormalities in VWD that distinguish VWD from hemophilia A are decreased VWF antigen level, and decreased VWF activity, often measured using the ristocetin cofactor activity assay and a prolonged closure time using the platelet function analyzer PFA-100. In type III VWD, factor VIII levels may be very low (<5 percent of normal), making it difficult to distinguish from classical hemophilia. The lack of a sex-linked pattern of inheritance in the family will help in the differential diagnosis.
Another variant of VWD that is particularly difficult to distinguish from hemophilia A is VWD-Normandy, in which VWF multimers are normal but plasma factor VIII levels are low.41 Several mutations causing VWD-Normandy have been described, but all of them result in decreased binding of factor VIII to VWF.42 The result is shortening of the intravascular survival of factor VIII and thus reduced factor VIII activity. The Normandy variant of VWD should be suspected in patients with mild hemophilia A who do not exhibit a sex-linked recessive inheritance pattern.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


