General Overview and Incidence
Overview of Hemophilia
The hemophilias are bleeding disorders caused by the absence or decrease of factor VIII in hemophilia A or factor IX in hemophilia B.
Coagulation
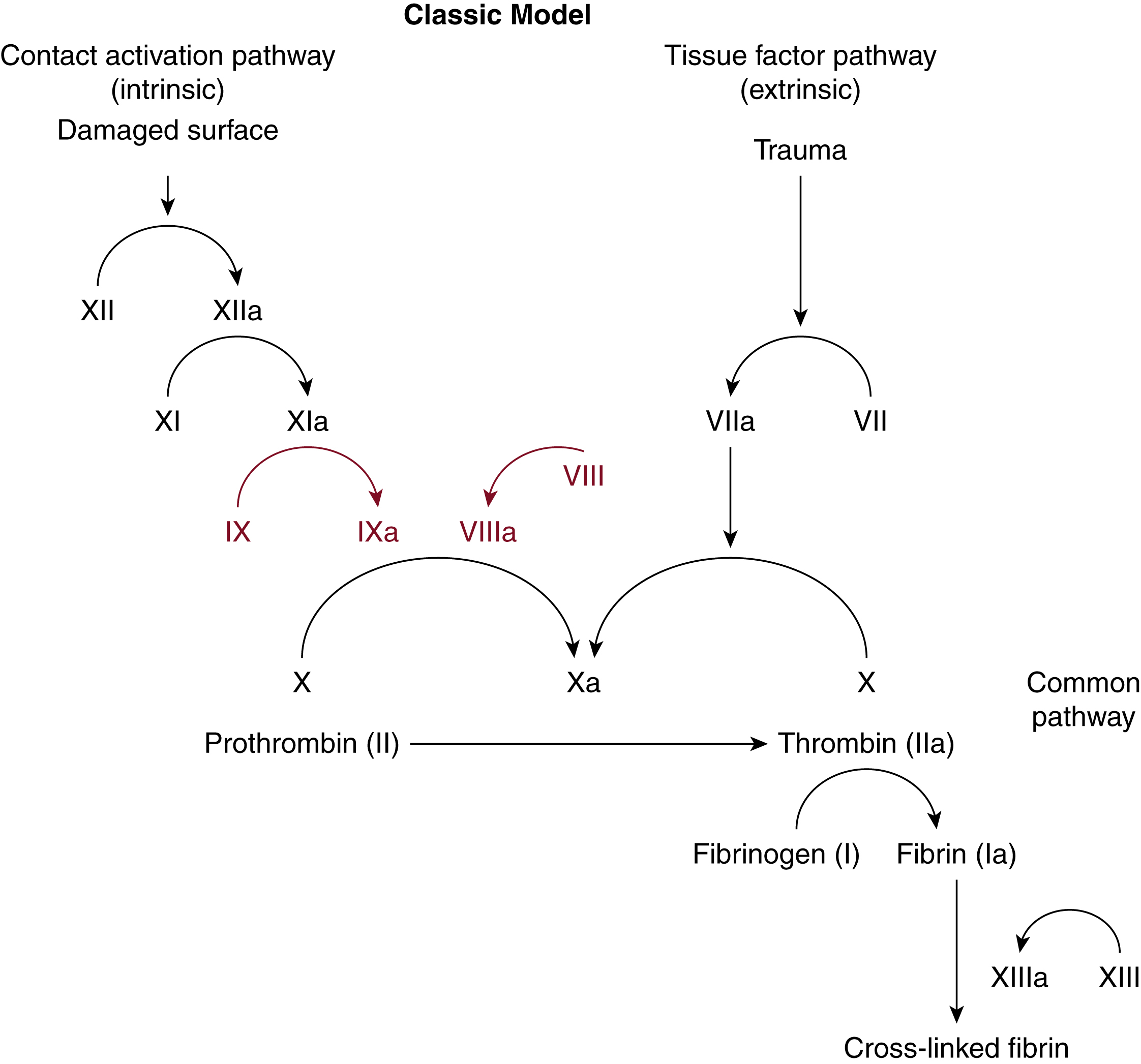
Factors VIII and IX (FVIII and FIX) are part of the coagulation cascade of secondary hemostasis. Classically, the coagulation cascade has two initial pathways leading to fibrin formation ( Fig. 18.1 ). These are the contact activation or intrinsic pathway, and the tissue factor or extrinsic pathway. The pathways are a series of reactions in which a zymogen of a serine protease is activated, which then catalyzes the next reaction in the cascade, ultimately resulting in cross-linked fibrin.
Roles of Factors VIII and IX in Coagulation
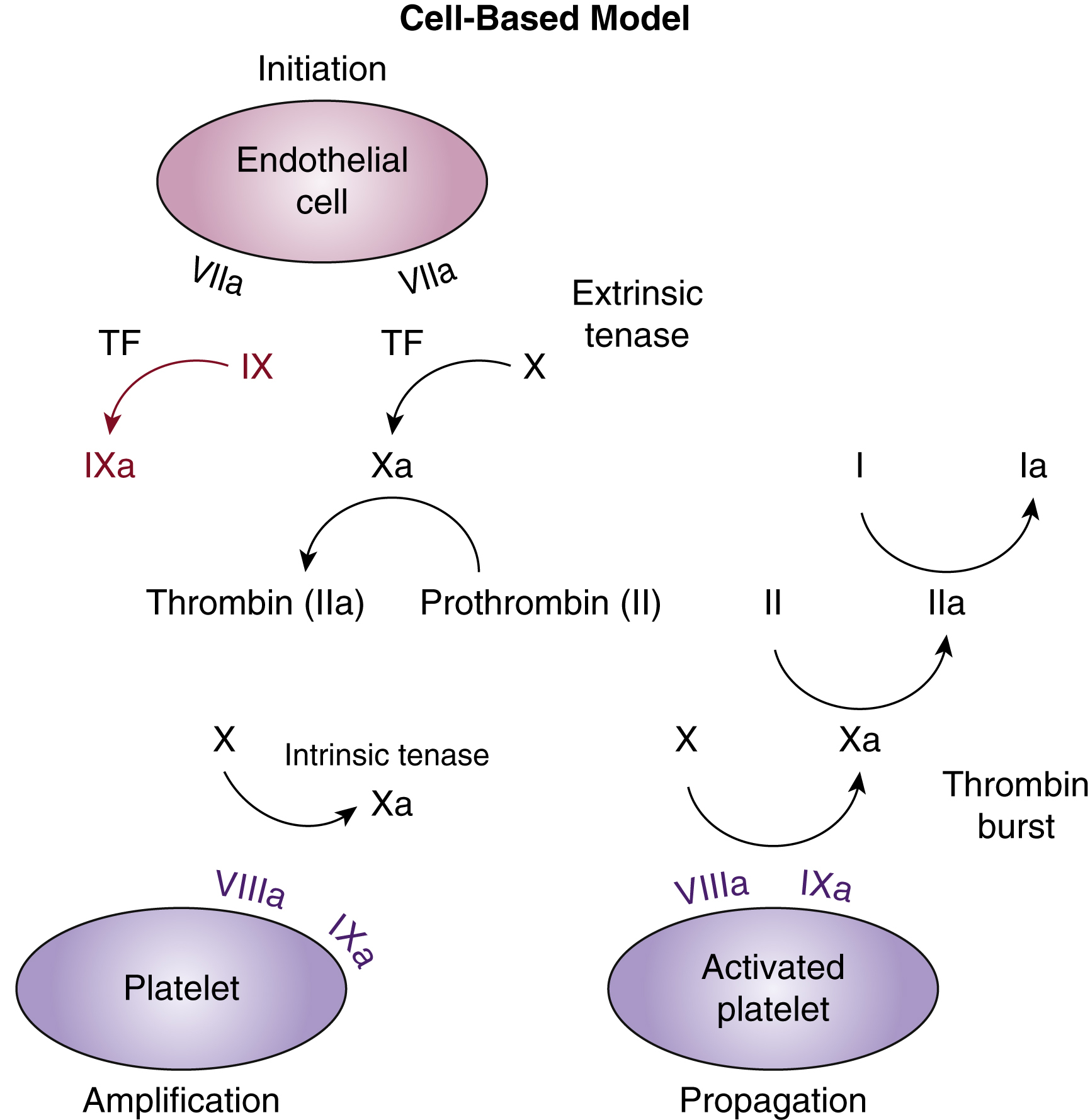
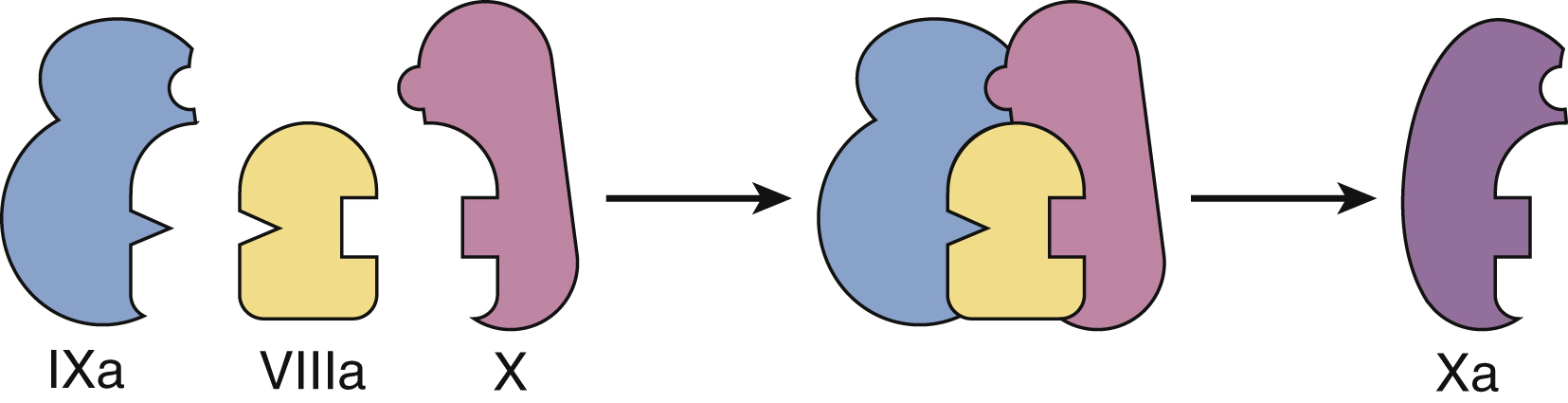
In vivo the coagulation cascade takes place on the surface of endothelial cells and platelets ( Fig. 18.2 ). The primary driver of secondary hemostasis is the factor VII or tissue factor pathway, which activates factor X to provide an initial thrombin burst. This is followed by amplification of factor Xa production by the complex of activated FVIII and FIX ( Fig. 18.3 ). Factor Xa produced by the factor VIIIa and IXa “tenase” complex, which also consists of calcium and phospholipids, then generates large amounts of thrombin and drives fibrin deposition and hemostasis.
Incidence and Inheritance
Incidence
Hemophilia A occurs in approximately 1:5000 males, whereas hemophilia B occurs less frequently, in about 1:20,000 males. Inheritance patterns are not affected by race or geography. There are more than 17,000 affected people in the United States, and it is estimated that more than 400,000 people are affected worldwide. Many affected individuals are from resource-poor nations that do not report hemophilia cases.
Inheritance
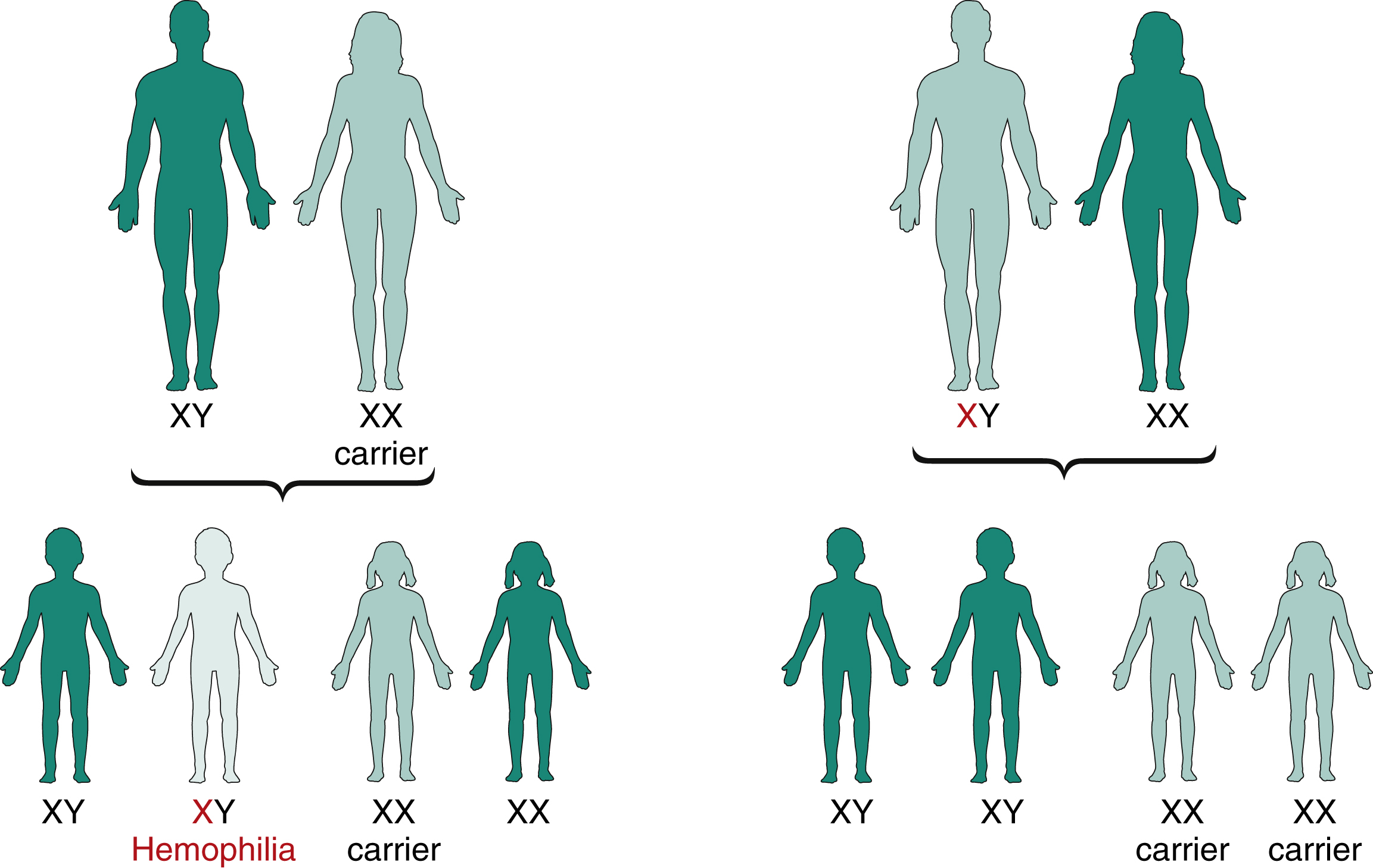
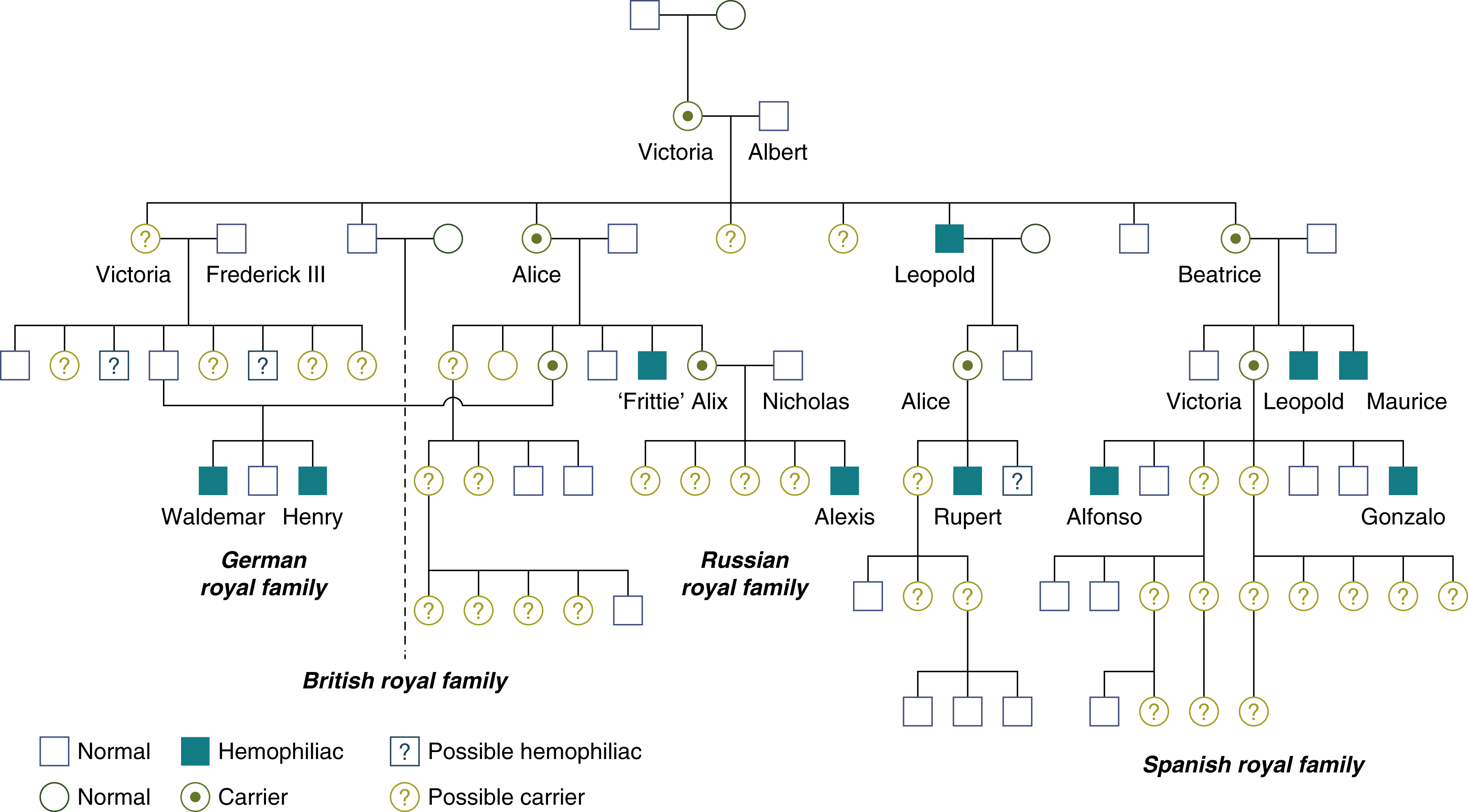
Both FVIII and FIX are located on the X chromosome and so are passed on in an x-linked pattern ( Fig. 18.4 ). Up to 30% of cases do not have a positive family history. The inherited nature of hemophilia was recognized in the Talmud of the second century, which read, “If she circumcised her first son and he died, and her second son and he too died, she should not circumcise her third son.” More recently, one of the most famous hemophilia pedigrees is that of the “royal disease” of the house of Queen Victoria, whereby most of the royal houses of Europe carried hemophilia B ( Fig. 18.5 ).
Etiology and Histopathology
Factor Level Determines Bleeding Phenotype
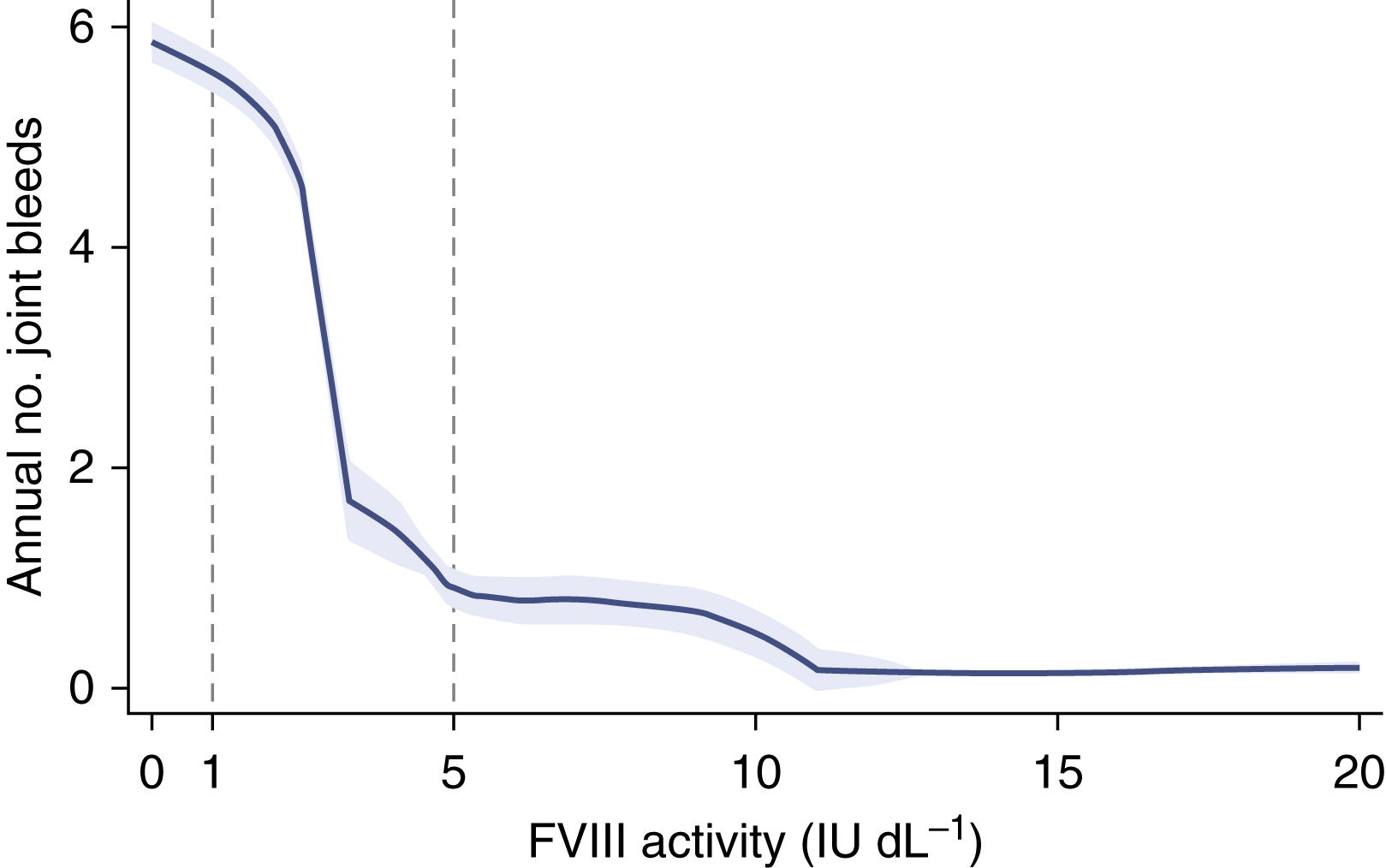
The baseline factor level of factor VIII or IX determines the severity of the bleeding symptoms ( Table 18.1 ). Lower baseline factor level correlates with bleeding rate ( Fig. 18.6 ). Although bleeding can occur at any location, hemorrhage into muscles or joints with little or no obvious trauma is the hallmark of hemophilia.
| Factor Activity (VIII or IX) | Phenotype |
|---|---|
| <1% activity | “Severe” hemophilia with spontaneous bleeding |
| 1% to 5% | “Moderate” disease with occasional spontaneous bleeding, severe bleeding with any surgery or trauma |
| >5% but <40% | “Mild” disease, rare spontaneous bleeding, severe bleeding if challenged with major surgery or trauma |
Molecular Basis of Hemophilia A and B
Common Mutations
Almost half of hemophilia A mutations involve inversion of exon 22. The remainder of mutations are primarily missense, frameshift, and nonsense mutations. Hemophilia B mutations are about half missense mutations, with the remainder mostly nonsense and frameshift mutations.
Inversion
Inversion 22 is the most common mutation in hemophilia A ( Fig. 18.7 ). It occurs through intrachromosomal recombination between a portion of exon 22 and distal reverse-homologous elements. A database is maintained of reported hemophilia mutations at http://www.factorviii-db.org/ .

Inhibitors in Hemophilia
Incidence and Etiology
Inhibitors are anti-factor antibodies that develop owing to exposure to exogenous “foreign” FVIII or FIX protein. Factor VIII antibodies are usually neutralizing antibodies, inhibiting the function of FVIII, whereas FIX antibodies typically cause anaphylaxis. Factor VIII antibodies occur in up to 20% to 25% of patients, whereas factor IX antibodies occur in 2% to 5% of patients. The greatest risk for developing antibodies is during the first 50 exposures to factor products. Some FVIII antibodies are of low titer and may spontaneously resolve, whereas higher titer antibodies will not. Bethesda assay (discussed in the section on acquired FVIII deficiency) is used to evaluate the strength of the inhibitor and response to treatment.
Molecular Basis
Association with mutations
The majority of mutations associated with inhibitors in both hemophilia A and B are missense mutations resulting in truncated proteins.
Immunology
The combination of a predisposing FVIII mutation and a variable response from human leukocyte antigen (HLA) class II are the main contributors to the risk of development of antibodies. Lower-risk mutations and HLA class II combinations are unlikely to develop antibodies to infused factor products, whereas higher-risk combinations may lead to antibody development. T-regulatory cells and other immune regulatory molecules then modulate the final immune response.
Acquired FVIII Deficiency
Acquired factor inhibitors are autoantibodies that arise spontaneously in patients without inherited coagulation defects. Inhibitors to FVIII are the most common acquired inhibitor. Estimates are 2 to 4 cases per 1 to 2 million persons, and a higher incidence of cases is found in postpartum women and the elderly (>60 years). Typically the diagnosis is made from a factor deficiency and a prolonged partial thromboplastin time (PTT) that does not correct with 1:1 mixing of patient plasma and normal pooled plasma (NPP). Quantitation of the inhibitor titer by Bethesda assay is used to monitor patients. One Bethesda unit is defined as the amount of inhibitor that decreases the residual FVIII in NPP by 50%. Different dilutions are necessary to identify the reaction of residual activity of 50%. Low-titer antibodies are found in lower dilutions and high-titer antibodies are found in higher dilutions. Patients with Bethesda titers below 5 can be treated with high-dose FVIII, whereas treatment for patients with Bethesda titers above 5 requires bypassing agents such as factor VIIa (NovoSeven) or factor eight inhibitor bypassing activity (FEIBA). Immunosuppression using prednisone, cyclophosphamide, and rituximab is used in conjunction with bypassing agents to eliminate the inhibitor.
Clinical Features
Bleeding Phenotypes
The severity of bleeding is determined by baseline factor level (see Fig. 18.6 ). Patients with factor levels greater than 12% rarely have spontaneous bleeding. This is important therapeutically because a small increment in factor level can dramatically change the bleeding phenotype. Typical spontaneous bleeding is into the large joints: knees, ankles, and elbows.
Bleeding Emergencies
Bleeding in hemophilia may be life-threatening depending on the location and must be treated emergently. Intracranial hemorrhage (ICH) is the leading cause of death in individuals with hemophilia. ICH can occur spontaneously or after trauma and may be a presenting sign in the newborn period. ICH may present with only a headache, so a high degree of clinical suspicion is needed. Iliopsoas muscle hematomas may become large enough to threaten exsanguination and are life-threatening bleeds. Bleeding into the calf or forearm may cause compartment syndrome and thus also requires emergency treatment.
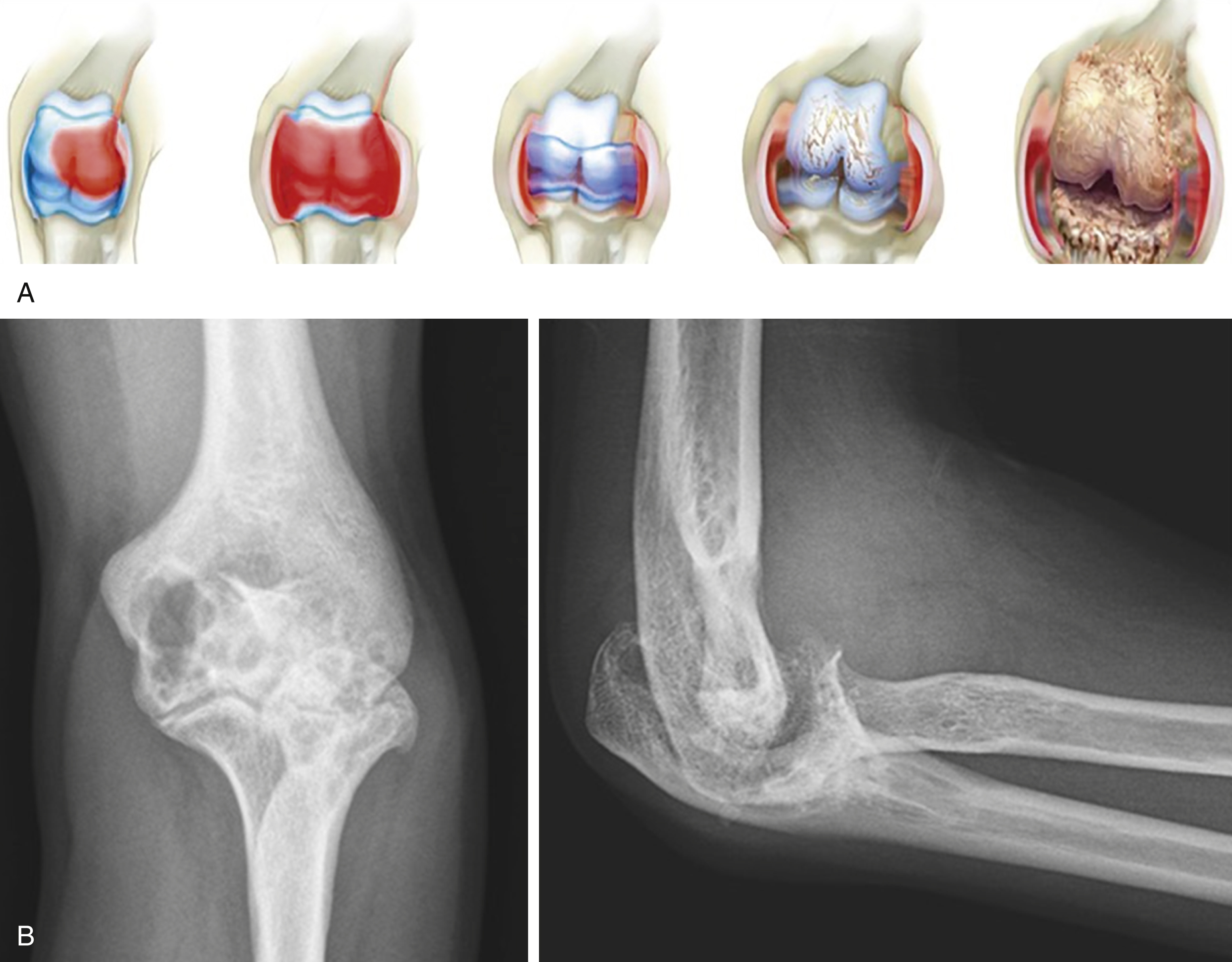
Joint Disease Progression
Recurrent bleeding in a joint leads to a progression of joint tissue injury that induces synovial growth and eventually cartilage and bone destruction ( Fig. 18.8 ). It is estimated that only a few bleeding episodes into the same joint without treatment will lead to significant damage of the joint. By definition a “target joint” has 4 recurrent bleeds over a 6-month period.