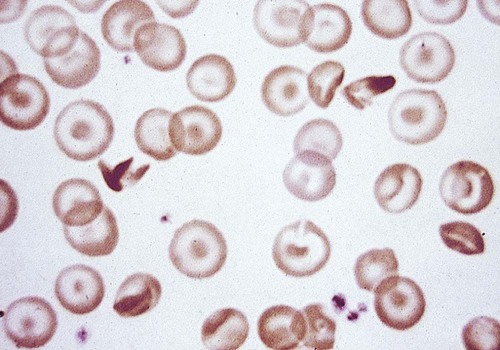
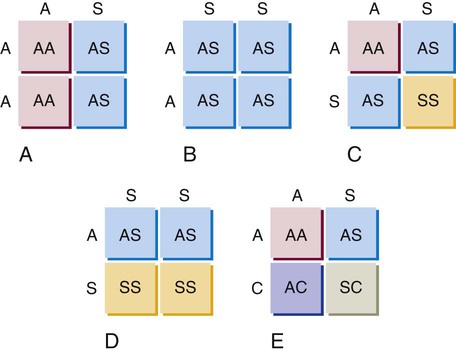
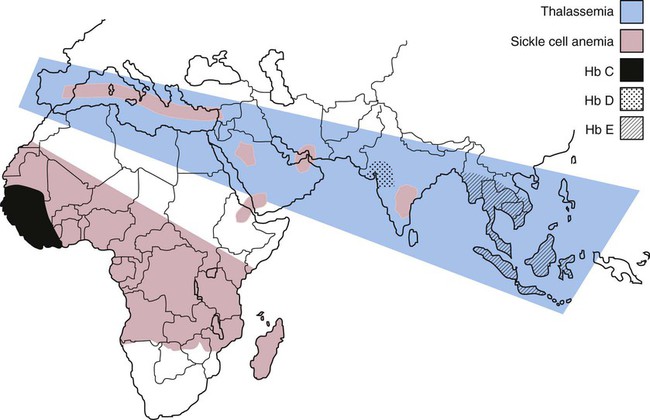
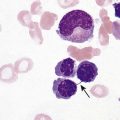
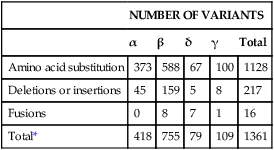
After completion of this chapter, the reader will be able to: 1. Compare and contrast structural hemoglobin (Hb) disorders with thalassemias. 2. Briefly describe globin gene structure and the development of normal human hemoglobins throughout prenatal and postnatal life. 3. Characterize the categories of the molecular abnormalities found in the various hemoglobinopathies. 4. Differentiate between homozygous and heterozygous states. 5. Define the pathologic basis of sickle cell disorder. 6. Describe the inheritance pattern of Hb S. 7. State the amino acid substitution found in Hb S and describe the electrophoretic mobility of Hb S. 8. Describe the solubility of Hb S in the deoxygenated state. 9. Discuss the clinical presentation of sickle cell disease, including differentiation among types of sickle cell crises. 10. Locate the geographic region, define the frequency of occurrence, and describe the impact of the incidence of Plasmodium falciparum malaria and glucose-6-phosphate dehydrogenase deficiency on hemoglobin variants. 11. Describe the peripheral blood cell analysis, chemistry panels, and other laboratory procedures used in the diagnosis of hemoglobinopathies. 12. Define the treatment goal for Hb S disease and discuss various treatments and their purposes. 13. Identify the amino acid substitution and electrophoretic mobility of Hb C and Hb C-Harlem. 14. Describe the amino acid substitution in Hb E and the importance of genetic counseling. 15. Explain the importance of and methods for differentiating Hb D and Hb G from Hb S. 16. Describe the laboratory findings in patients who are compound heterozygotes for Hb S and Hb D, Hb O-Arab, Hb Korle Bu, or Hb C-Harlem. 17. Identify the causes of methemoglobinemia. 18. Describe the inheritance patterns and causes of unstable hemoglobins. 19. Discuss hemoglobins with increased and decreased oxygen affinities, and explain how they differ from unstable hemoglobins. 20. Predict possible inheritance of hemoglobinopathies from the genotype of parents and the inheritance pattern for the gene. 21. Interpret patient hemoglobin electrophoresis patterns on cellulose acetate or agarose gel at pH 8.4 and on citrate agar at pH 6.0 to 6.2 when given control patterns. 22. Correlate hemoglobin electrophoresis and solubility test results to identify hemoglobinopathies. A typical field in the patient’s peripheral blood film is shown in Figure 26-1. Electrophoresis on cellulose acetate at alkaline pH showed 50.9% Hb S and 49.1% Hb C. 1. Select confirmatory tests that should be performed and describe the expected results. 2. Describe the characteristic RBC morphology on the peripheral blood film. 3. Based on the electrophoresis and RBC morphology results, what diagnosis is suggested? 4. If this patient were to marry a person of genotype Hb AS, what would be the expected frequency of genotypes for each of four children? Hemoglobinopathy refers to a disease state (“opathy”) involving the hemoglobin (Hb) molecule. All hemoglobinopathies result from a genetic mutation in one or more genes that affect hemoglobin synthesis. The genes that are mutated can code for either the proteins that make up the hemoglobin molecule (globin chains) or the proteins involved in synthesizing or regulating synthesis of the globin chains. Regardless of the mutation encountered, all hemoglobinopathies affect hemoglobin synthesis in one of two ways: qualitatively or quantitatively. In qualitative hemoglobinopathies, hemoglobin synthesis occurs at a normal or near-normal rate, but the hemoglobin molecule has an altered amino acid sequence within the globin chains. This change in amino acid sequence alters the structure of the hemoglobin molecule (structural defect) and its function (qualitative defect). In contrast, thalassemias result in a reduced rate of hemoglobin synthesis (quantitative) but do not affect the amino acid sequence of the globin chains. A reduction in the amount of hemoglobin synthesized produces an anemia and stimulates the production of other hemoglobins not affected by the mutation in an attempt to compensate for the anemia. Based on this distinction, hematologists divide hemoglobinopathies into two categories: structural defects (qualitative) and thalassemias (quantitative). To add confusion to the classification scheme, many hematologists also refer to only the structural defects as hemoglobinopathies. This chapter describes the structural or qualitative defects that are referred to as hemoglobinopathies; the quantitative defects (thalassemias) are described in Chapter 27. As discussed in Chapter 10, there are six functional human globin genes located on two different chromosomes. Two of the globin genes, α and ζ, are located on chromosome 16 and are referred to as α-like genes. The remaining four globin genes, β, γ, δ, and ε, are located on chromosome 11 and are referred to as β-like genes. In the human genome, there is one copy of each globin gene per chromatid for a total of two genes per person with the exception of α and γ. There are two copies of the α and γ genes per chromatid for a total of four genes per person. Each globin gene codes for the corresponding globin chain: the α-globin gene is used as the template to synthesize the α-globin chain, the β-globin gene codes for the β-globin chain, and so forth. More than 1000 structural hemoglobin variants (hemoglobinopathies) are known to exist throughout the world, and more are being discovered regularly (Table 26-1).1 Each of these hemoglobin variants results from one or more genetic mutations that alter the amino acid sequence. Some of these changes alter the molecular structure of the hemoglobin molecule, ultimately affecting hemoglobin function. The types of genetic mutations that occur in the hemoglobinopathies include point mutations, deletions, insertions, chain extensions, and fusions involving one or more of the adult globin genes—α, β, γ, and δ.2 TABLE 26-1 Molecular Abnormalities of Hemoglobin Variants *The table is designed to provide a relative distribution of mutation types and includes thalassemias and structural variants. Mutations are being added regularly, and at the time of publication there were 1034 hemoglobin variants and 395 thalassemias. The total number of entries in the table (1361) is less than the sum of the hemoglobin variants and thalassemias because some mutants are listed in both categories. Data from Patrinos GP, Giardine B, Riemer C, et al: Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies, Nucl Acids Res 32(database issue):D537-541, 2004. Available at: http://globin.cse.psu.edu/hbvar/menu.html. Accessed May 3, 2010. Point mutation is the most common type of genetic mutation occurring in the hemoglobinopathies. Point mutation is the replacement of one original nucleotide in the normal gene with a different nucleotide. Because one nucleotide is replaced by one nucleotide, the codon triplet remains intact, and the reading frame is unaltered. This results in the substitution of one amino acid in the globin chain product at the position corresponding to the location of the original point mutation. As can be seen in Table 26-1, 1128 of the 1361 known hemoglobin variants result from a point mutation that causes an amino acid substitution. It also is possible to have two point mutations occurring in the same globin gene, which results in two amino acid substitutions within the same globin chain. Over 30 mutations occur by this mechanism.2 Deletions involve the removal of one or more nucleotides, whereas insertions result in the addition of one or more nucleotides. Usually deletions and insertions are not divisible by three and disrupt the reading frame, which leads to the nullification of synthesis of the corresponding protein. This is the case for the quantitative thalassemias (see Chapter 27). In hemoglobinopathies, the reading frame usually remains intact, however; the result is the addition or deletion of one or more amino acids in the globin chain product, which sometimes affects the structure and function of the hemoglobin molecule. Of the 1361 variants described in Table 26-1, 217 variants result from deletions or insertions or both.2 Gene fusions occur when two normal genes break between nucleotides, switch positions, and anneal to the opposite gene. For example, if a β gene and a δ gene break in similar locations, switch positions, and reanneal, the resultant genes would be βδ and δβ fusion genes in which the head of the fusion gene is from one original gene and the tail is from the other. As long as the reading frames are not disrupted and the globin chain lengths are similar, the genes are transcribed and translated into hybrid globin chains. The fusion chains fold differently, however, and affect the corresponding hemoglobin function. Sixteen fusion globin chains have been identified (see Table 26-1). Fishleder and Hoffman3 divided the structural hemoglobins into four groups: (1) abnormal hemoglobins that result in hemolytic anemia, such as Hb S and the unstable hemoglobins; (2) abnormal hemoglobins that result in methemoglobinemia, such as Hb M; (3) hemoglobins with either increased or decreased oxygen affinity; and (4) abnormal hemoglobins with no clinical or functional effect. Imbalanced chain production also may be associated in rare instances with a structurally abnormal chain, such as Hb Lepore,1 because of the reduced production of the abnormal chain. The functional classification of selected hemoglobin variants is summarized in Box 26-1. Many of the variants are clinically insignificant because they do not show any physiologic effect. As discussed previously, most clinical abnormalities are associated with the β chain followed by the α chain. Involvement of the γ and δ chains does occur, but because of the small amount of hemoglobin involved, it is rarely detected and is usually of no consequence. Box 26-2 lists clinically significant abnormal hemoglobins. The most frequently occurring of the abnormal hemoglobins and the most severe is Hb S. As hemoglobins were reported, they were designated by letters of the alphabet. Normal adult hemoglobin and fetal hemoglobin were called Hb A and Hb F. By the time the middle of the alphabet was reached, however, it became apparent that the alphabet would be exhausted before all mutations were named. Currently, some abnormal hemoglobins are assigned a common designation and a scientific designation. The common name is selected by the discoverer and usually represents the geographic area where the hemoglobin was identified. A single capital letter is used to indicate a special characteristic of the hemoglobin variants, such as hemoglobins demonstrating identical electrophoretic mobility but containing different amino acid substitutions, as in Hb G-Philadelphia, Hb G-Copenhagen, and Hb C-Harlem. The variant description also can involve scientific designations that indicate the variant chain, the sequential and the helical number of the abnormal amino acid, and the nature of the substitution. The designation [β6 (A3) Glu→Val] for the Hb S mutation indicates the substitution of valine for glutamic acid in the A helix in the β chain at the sixth position.1 Although the origin of sickle cell anemia has not been identified, symptoms of the disease have been traced in one Ghanaian family back to 1670.4 Sickle cell anemia was first reported by a Chicago cardiologist, Herrick, in 1910 in a West Indian student with severe anemia. In 1917, Emmel recorded that sickling occurred in nonanemic patients and in patients who were severely anemic. In 1927, Hahn and Gillespie described the pathologic basis of the disorder and its relationship to the hemoglobin molecule. These investigators showed that sickling occurred when a solution of red blood cells (RBCs) was deficient in oxygen and that the shape of the RBCs was reversible when that solution was oxygenated again.1,5 In 1946, Beet reported that malarial parasites were present less frequently in blood films from patients with SCD than in individuals without SCD.6 It was determined that sickle cell trait confers a resistance against infection with Plasmodium falciparum occurring early in childhood between the time that passively acquired immunity dissipates and active immunity develops.7 In 1949, Pauling showed that when Hb S is subjected to electrophoresis, it migrates differently than does Hb A. This difference was shown to be caused by an amino acid substitution in the globin chain. Pauling and coworkers defined the genetics of the disorder and clearly distinguished heterozygous sickle trait (Hb AS) from the homozygous state (Hb SS).1 As stated earlier, the genes that code for the globin chains are located at specific loci on chromosomes 16 and 11. The α-like genes (α and ζ) are located on the short arm of chromosome 16, whereas the β-like genes (β, γ, δ, and ε) are located on the short arm of chromosome 11. With the exception of the γ genes, which have four loci, each β-like gene has two loci. β-hemoglobin variants are inherited as autosomal codominants, with one gene inherited from each parent.1 Patients with SCD (Hb SS), Hb SC, or Hb S–β-thal have inherited a sickle (S) gene from one parent and an S, C, or β-thalassemia gene from the other. Among patients with SCD, individuals who are homozygotes (Hb SS) have more severe disease than individuals who are compound heterozygotes for Hb S (Hb SC or Hb S–β-thal). Heterozygotes (Hb AS) are generally asymptomatic. Using Hb S and Hb C as examples, Figure 26-2 illustrates the inheritance of abnormal hemoglobins involving mutations in the β gene. The highest frequency of the sickle cell trait (Hb AS) is in Africa, where the frequency is 20% to 40%.8 In the United States, approximately 12% of African Americans have a hemoglobin variant. The homozygous state (Hb SS) represents the most severe type of hemoglobinopathy, with an estimated incidence of 1 in 375 live African American births.9 The rate of occurrence of heterozygous sickle cell trait is 8% among African Americans, with the serious compound heterozygous states of Hb SC and Hb S–β-thal occurring at rates of 1 in 835 (0.12%) and 1 in 1667 (0.06%), respectively.8 Although in the United States SCD is found mostly in individuals of African descent, it also has been found in individuals from the Middle East, India, and the Mediterranean area (Figure 26-3). SCD can also be found in individuals from the Caribbean and Central and South America.10 The sickle cell mutation is becoming more prominent in southern India, particularly in certain tribes.11 Hb S is defined by the structural formula α2β26Glu→Val, which indicates that on the β chain at the sixth position, glutamic acid is replaced by valine. Glutamic acid has a net charge of (−1), whereas valine has a net change of (+0). This amino acid substitution produces a change in charge of (+1), which affects the electrophoretic mobility of the hemoglobin molecule. This amino acid substitution also affects the way the hemoglobin molecules interact with one another within the erythrocyte cytosol. The nonpolar (hydrophobic) valine amino acid has been placed in the position that the polar glutamic acid once held. Because glutamic acid is polar, the β chain folds in such a way that glutamic acid extends outward from the surface of the hemoglobin tetramer to bind water and contribute to hemoglobin solubility in the cytosol. Therefore, the hydrophobic valine is also extended outward, but instead of binding water it seeks a hydrophobic niche with which to bind. When Hb S is fully oxygenated, the quaternary structure of the molecule does not produce a hydrophobic pocket for valine to bind to, which allows the hemoglobin molecules to remain soluble in the erythrocyte cytosol like Hb A and maintains the normal biconcave disc shape of the RBCs. However, the natural allosteric change that occurs upon deoxygenation creates a hydrophobic pocket in the area of phenylalanine 85 and leucine 88, which allows the valine from an adjacent hemoglobin molecule to bind. This hemoglobin pairing creates an orientation that helps other hemoglobin molecules to form amino acid bonds and become the seed for polymer formation. Other hemoglobin pairs polymerize, forming a hemoglobin core composed of four hemoglobin molecules that elongate in a helical formation. An outer layer of 10 hemoglobin molecules forms around the four-hemoglobin-molecule core, creating the long, slender Hb S polymer.12–15 Hb S molecules within the RBCs become less soluble, forming tactoids or liquid crystals of Hb S polymers that grow in length beyond the diameter of the RBC causing sickling. In homozygotes, the sickling process begins when oxygen saturation decreases to less than 85%. In heterozygotes, sickling does not occur unless the oxygen saturation of hemoglobin is reduced to less than 40%.16 The blood becomes more viscous when polymers are formed and sickle cells are created.16 Increased blood viscosity and sickle cell formation slow blood flow. In addition to a decrease in oxygen tension, there is a reduction in the pH and an increase in 2,3-biphosphoglycerate. Reduced blood flow prolongs the exposure of Hb S–containing erythrocytes to a hypoxic environment, and the lower tissue pH decreases the oxygen affinity, which further promotes sickling. The end result is occlusion of capillaries and arterioles by sickled RBCs and infarction of surrounding tissue. Sickle cells occur in two forms: reversible sickle cells and irreversible sickle cells.17 Reversible sickle cells are Hb S–containing erythrocytes that change shape in response to oxygen tension. Reversible sickle cells circulate as normal biconcave discs when fully oxygenated but undergo hemoglobin polymerization, show increased viscosity, and change shape on deoxygenation. The vasoocclusive complications of SCD are thought to be due to reversible sickle cells that are able to travel into the microvasculature in the biconcave disk conformation due to their normal rheologic properties when oxygenated and then become distorted and viscous as they become deoxygenated, converting to the sickle cell configuration in the vessel. Not only the oxygen tension but also the level of intracellular hydration affects the sickling process. When RBCs containing Hb S are exposed to a low oxygen tension, hemoglobin polymerization occurs. Polymerized deoxyhemoglobin S activates a membrane channel called Psickle that is otherwise inactive in normal RBCs. These membrane channels open when the blood partial pressure of oxygen decreases to less than 50 mm Hg. Open Psickle channels allow the influx of Ca2+, raising the intracellular calcium levels and activating a second membrane channel called the Gardos channel. An activated Gardos channel causes the efflux of K+, which stimulates the efflux of Cl– through another membrane channel to maintain charge equilibrium across the RBC membrane. The efflux of these ions leads to water efflux and intracellular dehydration, effectively increasing the intracellular concentration of Hb S and intensifying polymerization. Another contributor to K+ and Cl– efflux and the resultant dehydration is the K+/Cl– cotransporter system. Ironically, this system is activated by dehydration and positively charged hemoglobins such as Hb S and Hb C. The K+/Cl– cotransporter pathway is also activated by the low pH encountered in the spleen and kidneys. One potential explanation for the altered function of the membrane channels is oxidative damage triggered by Hb S polymerization. Injury to the RBC membrane induces adherence to endothelial surfaces, which causes RBC aggregation, produces ischemia, and exacerbates Hb S polymerization.7 Another important factor in the pathophysiology of SCD involves the redistribution of phospholipids in the RBC membrane, which contributes to hemolysis, vasoocclusive crisis, stroke, and acute chest syndrome. In the membranes of normal RBCs, choline phospholipids like sphingomyelin and phosphatidylcholine are located on the outer surface, whereas aminophospholipids like phosphatidylserine (PS) and phosphatidylethanolamine are primarily on the inner portion of the membrane. This asymmetrical distribution of membrane phospholipids is accomplished by adenosine triphosphate–dependent enzymes called translocases or flippases. Inhibition of flippases and activation of an enzyme called scramblase cause a more random distribution of membrane phospholipids, which increases the number of choline phospholipids on the interior half of the membrane and the number of aminophospholipids on the exterior membrane surface. The sickle cells of homozygotes (Hb SS) express 2.1% PS on erythrocyte exterior surfaces compared with 0.2% for normal Hb AA controls.18,19 It is hypothesized that Hb S polymerization may produce microparticles and iron complexes that adhere to the RBC membrane and generate reactive oxygen species, which, along with increased intracellular calcium or protein kinase C activation, may contribute to flippase inhibition and scramblase activation.20,21 PS on the exterior surface of RBCs binds thrombospondin on vascular endothelial cells,22 enhancing adherence between RBCs and the vessel wall and contributing to vasoocclusive crisis, activation of coagulation, and decreased RBC survival.23,24 In addition, RBCs with PS on the external membrane surface are vulnerable to hydrolysis by secretory phospholipase A2 (sPLA2), which generates lysophospholipids and fatty acids like lysophosphatidic acid. This results in vascular damage that contributes to acute chest syndrome.25,26 The clinical manifestations of SCD can vary from no symptoms to a potentially lethal state with the variety of symptoms listed in Box 26-3. Hundreds of hemoglobin variants are known; however, only four are clinically significant: Hb SS, Hb SC, Hb S–β0-thal, and Hb S–β+-thal. These four clinically severe forms have high morbidity and mortality rates. Average life span for Hb SS patients is 45 years; for Hb SC patients, it is 65 years, but life expectancy is increasing over time because of research findings and improved patient care.27 Individuals affected with SCD are characteristically symptom free until the second half of the first year of life because of the protective effect of Hb F.28 Toward the end of the first 6 months of life, mutated β chains begin to be produced and gradually replace normal γ chains, which causes Hb S levels to increase and Hb F levels to decrease. Erythrocytes containing Hb S become susceptible to hemolysis, and a progressive hemolytic anemia and splenomegaly may become evident.
Hemoglobinopathies (Structural Defects in Hemoglobin)
Case Study
Patient Results
Reference Range
WBCs (× 109/L)
11.9
4.5-11.0
RBCs (× 1012/L
3.67
4.00-5.40
Hb (g/dL)
10.9
12.0-15.0
Hct (%)
32.5
35-49
RDW (%)
19.5
11.5-14.5
Platelets (× 109/L)
410
150-450
Segmented neutrophils (%)
75
50-70
Lymphocytes (%)
18
18-42
Monocytes (%)
3
2-11
Eosinophils (%)
3
1-3
Basophils (%)
1
0-2
Reticulocytes (%)
3.1
0.5-1.5
Structure of Globin Genes
Genetic Mutations
NUMBER OF VARIANTS
α
β
δ
γ
Total
Amino acid substitution
373
588
67
100
1128
Deletions or insertions
45
159
5
8
217
Fusions
0
8
7
1
16
Total*
418
755
79
109
1361
Pathophysiology
Nomenclature
Hemoglobin S
Sickle Cell Anemia
History
Inheritance Pattern
Incidence
Etiology and Pathophysiology
Clinical Features