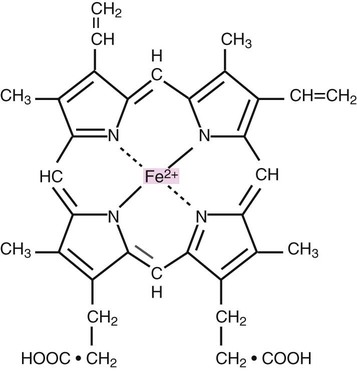
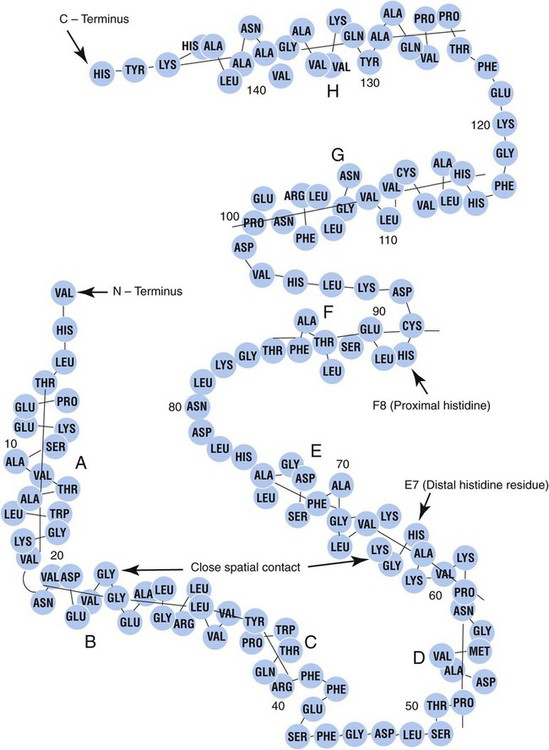
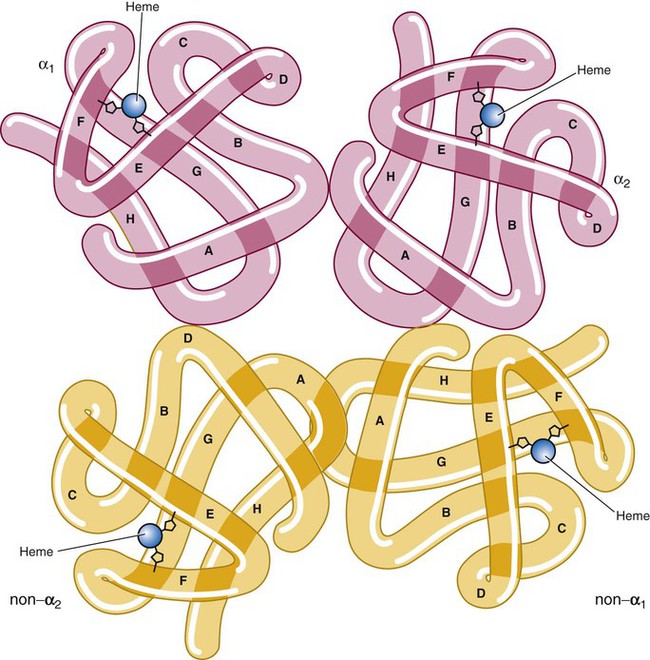
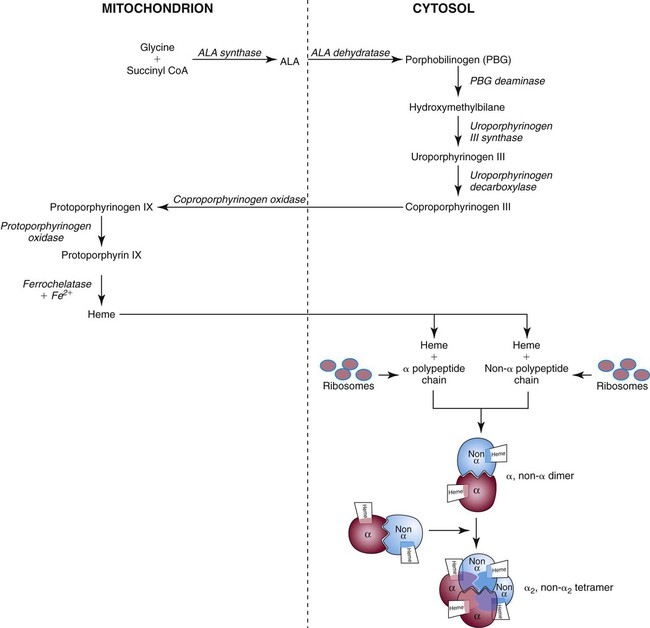
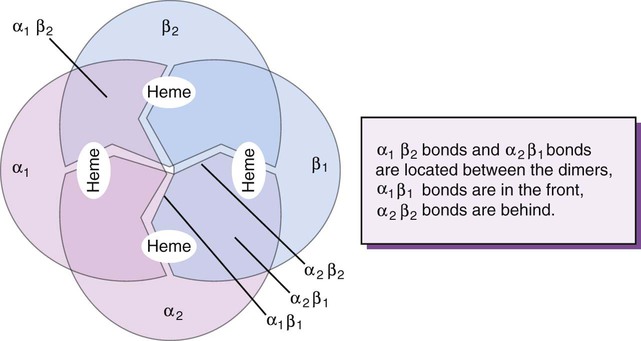
After completion of this chapter, the reader will be able to: 1. Describe the primary structure of the globin chains found in hemoglobin. 2. Describe the quaternary structure of hemoglobin. 3. Describe the biosynthesis of heme and globin. 4. Differentiate steps in heme synthesis that occur in the mitochondria and the cytoplasm. 5. Identify the ontogeny of hemoglobin with emphasis on the hemoglobin of newborns and adults. 6. Identify the three types of normal hemoglobin in adults and their reference intervals. 7. Describe the regulatory effects of hemoglobin metabolism. 8. Identify the important role that hemoglobin plays in maintaining body functions. 9. Describe the mechanism by which hemoglobin carries oxygen to the tissue. 11. Explain the significance of the sigmoid shape of the oxygen dissociation curve. 12. Correlate right and left shifts in the oxygen dissociation curve with conditions that can cause shifts in the curve. 13. Identify the P50 value (amount of oxygen needed to saturate 50% of hemoglobin) as it pertains to a normal oxygen dissociation curve. 14. Differentiate T and R forms of hemoglobin. 15. Identify the source of production of 2,3-bisphosphoglycerate and describe its impact on hemoglobin oxygenation. 16. Identify the oxygen affinity of fetal hemoglobin. 17. Compare and contrast the composition of the chemically modified hemoglobins—methemoglobin, carboxyhemoglobin, and sulfhemoglobin—and their affinity for oxygen. 18. Compare and contrast oxygenated, deoxygenated, and oxidized hemoglobin and ferric versus ferrous iron. 19. Describe how hemoglobin is routinely measured in the laboratory. 20. Identify how different kinds of hemoglobins are identified by laboratory tests. 21. Identify the gene locations of the globins that make up the hemoglobin molecule, including the number of genes for each globin chain (for Hb A, A2, and F) and their general arrangement on chromosomes. 1. Were these hemoglobin results within expected reference intervals? 2. Why were the mother’s and the newborn’s hemoglobin results so different? 3. What is the difference between a hemoglobin test and the hemoglobin electrophoresis test? 4. Why were the mother’s and newborn’s hemoglobin electrophoresis results so different? Hemoglobin (Hb) is the first protein whose structure was described using x-ray crystallography.1–4 The hemoglobin molecule is a conjugated globular protein consisting of four heme groups and two heterogenous pairs of polypeptide chains (Figure 10-1). Heme consists of a ring of carbon, hydrogen, and nitrogen atoms called protoporphyrin IX with an atom of divalent ferrous iron (Fe2+) attached (ferroprotoporphyrin, Figure 10-2). Each of the four heme groups is positioned in a pocket of the polypeptide chain near the surface of the hemoglobin molecule. Each heme molecule combines reversibly with one oxygen molecule. Owing to its double bonds, heme renders blood red. The four globin chains comprising each hemoglobin molecule consist of two identical pairs of unlike polypeptide chains, 141 to 146 amino acids each. Variations in amino acid sequences give rise to different types of polypeptide chains. Each chain is designated by a Greek letter (Table 10-1).5 TABLE 10-1 Each globin chain is divided into eight helices and seven nonhelical segments (Figure 10-3). The helices, designated A to H, contain subgroup numberings for the sequence of the amino acids in each helix and are relatively rigid and linear. The flexible nonhelical segments connect the helices, as reflected by their designations: NA for the sequence between the N-terminus and the A helix, AB between the A and B helices, and so forth with BC, CD, DE, EF, FG, GH, and finally HC between the H helix and the C-terminus. The quaternary structure of hemoglobin, also called a tetrameric molecule, describes the complete hemoglobin molecule. The complete hemoglobin molecule is spherical, has four heme groups attached to four polypeptide chains, and may carry four molecules of oxygen. It is composed of two α globin chains and two non-α globin chains. Each globin chain has a heme group attached. Each heme molecule is capable of carrying one molecule of oxygen. Strong α1,– non-α1 and α2,–non-α2 dimeric bonds hold the molecule in a stable form. Tetrameric α1–non-α2 and α2,–non-α1 bonds also contribute to the stability of the structure (Figure 10-4).6,7 Heme biosynthesis occurs in the mitochondria and cytoplasm of bone marrow RBC precursors, beginning with the pronormoblast (also known as proerythroblast) through the circulating polychromatic (also known as polychromatophilic) erythrocyte (see Chapter 8). As they lose their mitochondria and the citric/tricarboxylic acid cycle, mature RBCs can no longer make hemoglobin. Heme biosynthesis begins with the condensation of glycine and succinyl coenzyme A (CoA) catalyzed by aminolevulinate synthase (ALAS) to form aminolevulinic acid (ALA). ALA dehydratase (also known as ALA dehydrase, porphobilinogen synthase) in the presence of ALA catalyzes the formation of porphobilinogen. Porphobilinogen deaminase, also known as hydroxymethylbilane synthase, in the presence of porphobilinogen catalyzes the formation of hydroxyl methylbilane. This pathway continues until, in the final step of production of heme, Fe2+ combines with protoporphyrin IX in the presence of ferrochelatase/heme synthase to make heme (Figure 10-5).6 The production of globin chains takes place in RBC precursors from the pronormoblast through the circulating polychromatic (polychromatophilic) erythrocyte, but not in the mature RBC.8,9 Globin proteins arise via transcription of the genetic code to messenger ribonucleic acid (mRNA) and translation of mRNA to the globin polypeptide chain. A slight excess of α-globin mRNA is present in pronormoblasts (proerythroblasts); however, β-globin mRNA is translated more efficiently than α-globin mRNA. This results in synthesis of sets of chains in approximately equal amounts. When synthesized, the chains are released from the ribosomes in the cytoplasm.7
Hemoglobin Metabolism
Case Study
Hemoglobin Structure
Heme Structure
Globin Structure
Symbol
Name
No. of Amino Acids
α
Alpha
141
β
Beta
146
γA
Gamma A
146 (position 136: alanine)
γG
Gamma G
146 (position 136: glycine)
δ
Delta
146
ε
Epsilon
Unknown
ζ
Zeta
141
θ
Theta
Unknown
Complete Hemoglobin Molecule
Hemoglobin Biosynthesis
Heme Biosynthesis
Globin Biosynthesis
Hemoglobin Metabolism