PIDs classically are associated with significant morbidity and early mortality caused by life-threatening infections in the first months and years of life. Before the availability of HSCT, these infections invariably caused death in the vast majority of affected children. SCIDS is the most widely recognized manifestation of a PID. SCIDS is a genetically heterogeneous group of over 20 mutations in 13 genes identified to date that result in the common phenotype of impaired T-cell production and/or function (
Table 103.1). These mutations in varied target genes lead to impaired immunity as a consequence of absent T-cell function, but often defects in B-cell and natural killer (NK) cell number or function occur in parallel, as many of the genes affect all three lineages. However, the level of B- and NK-cell activity is highly variable. In rare cases, autoimmune phenomena are observed in these patients.
13 This is particularly noteworthy in children with Omenn syndrome, which is caused by mutations in genes that participate in rearrangements of T-cell receptors and VDJ regions of immunoglobulins. Mutations in these genes elicit a phenotype that mimics graft-versus-host-disease as a consequence of the expansion of T-cell clones that are reactive to self-antigens. There is also reduced peripheral and central tolerance in these patients, which contributes to the observed phenotype.
Preparative Regimen
The first transplant for SCIDS using an HLA-matched sibling bone marrow donor was performed in 1968.
14 In this case, no preparative regimen was administered to prevent graft rejection due to the absence of an immunologic barrier to engraftment. Thus, donor hematopoietic stem cells simply were infused and successful immune reconstitution followed. However, fatal graftversus-host disease (GVHD) often occurred in donor/recipient settings in which there was a major HLA disparity. By testing transplantation in animal models of SCIDS, it was determined that by using the technique of donor T-cell depletion, it was possible to eliminate GVHD after HLA-mismatched HSCT, if thorough T-cell depletion was accomplished.
15,
16 In addition,
after successful HSCT in the mouse, there was durable engraftment of the donor cells with restored host immunity. However, even in the earliest clinical reports of HSCT after T-cell depleted HLA-haploidentical HSCT for SCIDS, B-cell engraftment was quite poor, as most patients required long-term immunoglobulin replacement after HSCT.
In a cohort of 161 patients treated by HSCT at a single U.S. institution, Railey et al. reported the results in individuals with SCIDS who received no pre-transplant chemotherapy or GVHD prophylaxis.
17 Notably, only 16 (10%) of these patients had HLA-identical sibling donors, and thus most received T-cell depleted HLA-haploidentical marrow. In this clinical series, it
was observed that survival was highest among those patients who were transplanted before 3.5 months of age (94% compared to 70%,
P = 0.002). Of those who died, the majority (76%) experienced progressive viral infections that pre-dated HSCT. There were very few deaths related to GVHD or infections that were acquired after HSCT. Thus, this series suggested that no chemotherapeutic preparation is necessary in patients with SCIDS who are treated by HSCT shortly after birth. However, many patients require regular immunoglobulin (IVIG) infusions after successful HSCT for SCIDS due to absent engraftment of donor B-cells, particularly in the absence of a conditioning regimen before HLA-haploidentical related donor HSCT in recipients with B-cell positive SCIDS.
18,
19Long-term follow-up of patients showed mixed results regarding the stability of long-term donor T-cell engraftment after HLA-haploidentical HSCT in the absence of a conditioning regimen before HSCT. With up to 25 years follow-up in a report by Buckley, 11 of 19 patients had no evidence of decreased T-cell function or diminished T-cell repertoire diversity. However, as noted above, most patients lacked donor B-cell engraftment and thus received long-term replacement immunoglobulin therapy.
8 In another series that included survivors with up to 16 years follow-up, 35% of the patients had low T-cell antigen receptor excision circle (TREC) levels and a limited T-cell repertoire as evidenced by oligoclonality that was demonstrated in 27.5% of patients.
20 Based upon these observations, it has been suggested that the use of an ablative conditioning regimen before HSCT may be required to ensure long-term T-cell engraftment in the setting of HLA-haploidentical donors.
Whether improved T-cell engraftment translates into improved survival remains in question, however. Patel et al. retrospectively reviewed a series of 22 patients with SCIDS who were treated by HSCT at a single institution.
21 In this series, no statistical difference in overall survival was with respect to whether a preparative regimen was administered regardless of donor type. In addition, experience from a large European cohort treated between 1968 and 2005 showed that chemotherapeutic preparation did not improve overall survival in patients with SCIDS.
22 These authors argued that a conventional ablative chemotherapeutic preparation does not improve survival in patients with SCIDS, particularly in those who undergo HSCT in the first 31/2 months of life. Unfortunately, proceeding directly to HSCT early in life is not always feasible, as affected individuals often are not identified in the first several months of life or before they have developed serious infections. Thus, the decision of whether to use a conditioning regimen should hinge on several salient clinical features, including the patient’s age, the donor, and the baseline immunologic function with regard to resistance to donor engraftment. Also, recipients with SCIDS who have a T-/B+ lymphocyte profile appear to have better outcomes than recipients who lack B-cells.
23 The advent of pilot newborn screening programs by TREC screening from blood spots has the potential to mitigate the limitation of patient age.
24,
25,
26
Reduced Intensity Conditioning
To date, optimization of a conditioning regimen that might be safely applied in all patients with SCIDS has been elusive. In an attempt to reduce transplant-related morbidity and mortality, reduced intensity conditioning regimens that are not ablative and contain a diverse array of immunosuppressive agents such as fludarabine, busulfan (BU), etoposide, cyclophosphamide (CY), and low-dose radiation have been tested.
27,
28,
29 Current data suggest that reduced intensity regimens are most appropriate in recipients with T-cell deficiency syndromes and in unrelated donor HSCT.
30,
31 However, graft rejection associated with inadequate suppression of residual host immune function remains a concern, particularly in patients with T- and NK-cell function at baseline. Ultimately, the selection of a preparative regimen and its intensity will vary according to the level of retained immunity in the recipient, and with the degree of HLA matching in donor hematopoietic cells.
Donor Source
HLA-identical marrow HSCT has yielded very good results in SCIDS, with approximately 70% to 80% of patients surviving long-term
32 (
Fig. 103.1). Because most patients lack a HLA-ID sibling donor, alternative donor sources have been explored in clinical trials. Roifman et al. reviewed transplant outcomes after HLA-mismatched related donor HSCT compared to HLA-matched unrelated donor HSCT for SCIDS.
33 They reported that survival was superior in those receiving HLA-matched unrelated marrow. Specifically, patients who received a HLA-matched unrelated HSCT had better engraftment and immune reconstitution. They concluded that HLA-matched unrelated marrow transplantation is superior to HLA-mismatched related donor HSCT and should be considered as the first choice when a related HLA-identical donor is unavailable. In other patient series noted above, excellent results were observed after T-cell depleted HLA-haploidentical HSCT. Thus, the optimal donor source for HSCT remains a point of controversy.
More recently, the application of umbilical cord blood transplantation (UCBT) has been explored in SCIDS. Fernandes et al. recently compared results after HLA-mismatched related donor and unrelated donor UCBT in patients with SCIDS.
34 There were 175 HLA-mismatched donor HSCT recipients and 74 UCBT recipients in this retrospective cohort of patients with SCIDS or Omenn syndrome. The median follow-up was 58 and 83 months in each group, respectively. Most UCBT recipients received a myeloablative preparative regimen, whereas a larger fraction of the related donor recipients did not. UCBT recipients were more likely to have complete donor chimerism and faster lymphocyte recovery independent of the preparative regimen. T-cell engraftment was equivalent in the two groups, and immunoglobulin replacement was discontinued earlier after transplantation in the UCBT group. There was a statistically insignificant higher incidence of grade II—IV acute GVHD and statistically significant rate of chronic GVHD after UCBT. However, the 5-year overall survival rates were equivalent. Other data suggest that B-cell engraftment may be better after UCBT.
35 Although a randomized, prospective trial has not been performed, these data indicate that UCB appears to represent a suitable alternative donor source.
Late Effects and Future Directions
There are limited series of long-term follow-up after HSCT for SCIDS in which the follow-up period exceeds 10 years.
20,
21,
36 In several of these series, loss of T-cell diversity and autoimmune disorders was observed late after HSCT, although follow-up through 25 years after HSCT suggests improvement in the T-cell repertoire.
37 In addition, patients with adenosine deaminase deficiency in particular have experienced a high incidence of central nervous system abnormalities that include motor dysfunction, sensorineural hearing loss, and cognitive deficits that contribute to emotional and behavioral difficulties.
20 In addition, chronic infection by human papilloma virus also has been reported.
20 It is unclear which, if any of these, is related to pre-transplant conditioning and GVHD prophylaxis or to the underlying genetic defect. Finally, for those who lack a transplant donor, gene therapy is an alternative therapy for some patients, although development of this technology is ongoing.
38 A consensus group has discussed plans for future multicenter studies in North America, both retrospective and prospective, focused on collecting data from newborn screening and natural history sources.
39 The importance of collaboration in developing new protocols for these rare disorders
was illustrated best by recent data showing that long-term mortality rates for both SCIDS and non-SCIDS patients with a PID are higher than their age- and gender-matched controls in the general population.
40 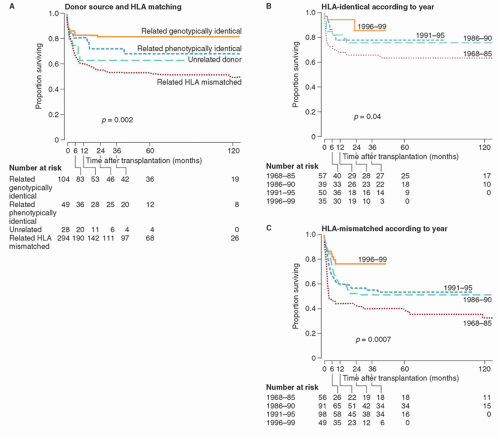
 Clinical Flow Cytometry
Clinical Flow Cytometry



