Helicobacter pylori and Gastric Neoplasms
Abha Goyal
INTRODUCTION
Helicobacter pylori, a Gram-negative bacterium, is the most important human pathogen of the Helicobacter genus. Studies by Warren and Marshal1 were the first to implicate H. pylori as the causative agent of chronic gastritis and ulcers. This discovery led to the Nobel Prize for Medicine and Physiology in 2005. H. pylori infection results in chronic injury of the stomach and chronic gastritis, which significantly increases the risk of gastric adenocarcinoma and mucosaassociated lymphoid tissue (MALT) lymphoma, generally described in the current World Health Organization (WHO) classification as extranodal marginal zone B-cell lymphoma.2, 3 and 4
In this chapter, we review the evidence supporting a role for H. pylori in gastric carcinogenesis, the stepwise pathologic changes of the gastric mucosa (gastritis, gastric atrophy, intestinal metaplasia, and dysplasia/adenoma) that underlie gastric cancer, the role of Helicobacter virulence factors in gastric cancer risk, the molecular mechanisms underlying H. pylori-associated gastric cancer, human genetic susceptibility factors including proinflammatory gene polymorphisms, and the potential contribution of bone marrow-derived stem cells in gastric carcinogenesis. We also review the evidence supporting a role for H. pylori in gastric lymphomagenesis, the mechanisms that may underlie H. pylori-associated MALT lymphoma, the molecular features of gastric MALT lymphomas, and the role of H. pylori eradication in the treatment of these low-grade lymphomas.
H. pylori AND GASTRIC CANCER
Evidence for a Role of H. pylori in Gastric Carcinogenesis
Gastric cancers are heterogeneous neoplasms that include several adenocarcinoma subtypes characterized by unique genetic and molecular features. This heterogeneity is likely a result of alternative underlying mechanisms of neoplastic development and progression. Since the early 1990s, much has been learned regarding the molecular basis and biological behavior of gastric adenocarcinoma and precursor lesions. The scientific breakthroughs that have led to and continue to propel our understanding of gastric carcinogenesis include the following: (a) the demonstration that H. pylori gastritis is associated with an increased risk of gastric cancer development, (b) the identification of a genetic basis for some familial forms of gastric cancer, and (c) the availability of robust molecular approaches that expand the traditional morphology-based characterization of gastric cancer subtypes.
The association of H. pylori infection and gastric adenocarcinomas led to the classification of H. pylori as a human carcinogen by a WHO panel in 1994,2 nearly a decade after the first report by Warren and Marshal1 implicating H. pylori as the bacteria that causes chronic gastritis. H. pylori infection appears to play a role predominantly during the initiating steps of gastric cancer, although the environment established by the ongoing inflammatory response to the infection may contribute to neoplastic progression.
Gastric cancers result from the action of several causal mechanisms and their interactions. The factors known to play a role in gastric cancer development include host genetic susceptibility factors, environmental exogenous factors that have carcinogenic activity such as dietary components and smoking, and the cellular injury that is caused by chronic gastritis (reviewed in Ref. [5]).
The evidence supporting a role for H. pylori in gastric carcinogenesis includes the following: (a) epidemiological studies reporting a close association between H. pylori and gastric cancer in several populations around the world, (b) characteristic histopathologic changes that mark the steps of gastric mucosal injury associated with gastric cancer risk (the Correa cascade), (c) animal models of H. pylori and gastric cancer, and (d) data showing that H. pylori eradication may reduce the risk of gastric cancer development.
H. pylori was initially classified as a human carcinogen based on the strong epidemiological association of H. pylori gastritis and gastric cancer.2,3 This association was explained by the fact that the majority of gastric cancers arise in a background of chronic gastritis, which is most commonly caused by H. pylori infection of the stomach.6,7 Overall, the risk of gastric cancer in patients with H. pylori gastritis is sixfold higher than that of people without H. pylori gastritis,8 and this risk increases for patients with more extensive degrees of gastric atrophy and intestinal metaplasia. Even for H. pylori-infected patients with nonatrophic gastritis, their gastric cancer risk is approximately twofold higher than that of healthy non-H. pylori-infected individuals. In addition, prospective studies demonstrated gastric cancer development in 2.9% of H. pylori-infected patients over a period of 7.8 years, but this incidence was higher in patients with extensive atrophic gastritis and intestinal metaplasia, reaching an incidence rate of 8.4% during a 10-year surveillance.9,10 Underscoring the importance of H. pylori in gastric carcinogenesis, the gastric cancer risk attributable to H. pylori infection has been estimated to be 75%.7
In addition, several animal models, including Mongolian gerbils and mice, develop gastritis as well as other H. pylori-associated gastric diseases, including intestinal metaplasia, dysplasia, and gastric cancer, following Helicobacter infection.11,12
Furthermore, H. pylori eradication reduced the incidence of gastric cancer in patients without atrophy and intestinal metaplasia at study baseline,13 and eradication of H. pylori infection in patients with early gastric cancer resulted in a decreased frequency of new cancers,14 supporting the notion that eradication may contribute to preventing progression from gastritis to gastric cancer.
Histologic Changes in the Progression of Chronic H. pylori Gastritis to Cancer
The chronicity associated with H. pylori gastritis is critical to the carcinogenic potential of the infection. H. pylori infection is generally acquired during childhood and persists throughout life unless the patient undergoes eradication treatment.15 Gastric cancer generally develops several decades after acquisition of the infection and is associated with the progression of mucosal damage during chronic gastritis.
Histologically, the progression of H. pylori-associated chronic gastritis to gastric cancer is characterized by a stepwise acquisition of mucosal changes, starting with chronic gastritis, the progressive damage of gastric glands resulting in mucosal atrophy, the replacement of normal gastric glands by intestinal metaplasia, and the development of dysplasia and carcinoma in some patients (Figs. 3-1 and 3-2). Gastritis is the first and longest lasting response to H. pylori infection of the stomach and is mediated by the activation of both humoral and cellular inflammatory responses within the gastric mucosa involving dendritic cells, macrophages, mast cells, the recruitment and expansion of T and B lymphocytes, plasma cells, and neutrophils.16 Despite the continued host inflammatory response, H. pylori is able to evade the host immune mechanisms and persist, causing chronic gastritis.
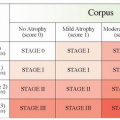
The pathologic changes of the gastric mucosa in which H. pylori-associated gastric cancer generally occurs may be described as atrophic gastritis, where, in a background of active chronic
inflammation, there is a progressive loss of the normal glandular structures of the gastric mucosa (gastric atrophy) and the replacement of the normal glandular epithelium by intestinal metaplasia. This stepwise series of histopathologic changes are named the Correa cascade. Given the association of gastric glandular atrophy and intestinal metaplasia and an increased risk of gastric cancer, these changes, in particular intestinal metaplasia, are accepted as a marker of gastric cancer risk and may be interpreted as preneoplastic epithelial changes in gastric carcinogenesis. Progression to gastric cancer is higher in patients with more extensive forms of atrophic gastritis with intestinal metaplasia involving large areas of the stomach, including the gastric body and fundus. This pattern of gastritis has been described as pangastritis or multifocal atrophic gastritis.6,17 Extensive gastritis involving the gastric body and fundus results in hypochlorhydria, allowing for bacterial overgrowth and increased carcinogenic activity in the stomach through the conversion of nitrites to carcinogenic N-nitroso compounds. H. pylori-associated pangastritis is frequently seen in the relatives of gastric cancer patients, which may contribute to gastric cancer clustering in some families.18
inflammation, there is a progressive loss of the normal glandular structures of the gastric mucosa (gastric atrophy) and the replacement of the normal glandular epithelium by intestinal metaplasia. This stepwise series of histopathologic changes are named the Correa cascade. Given the association of gastric glandular atrophy and intestinal metaplasia and an increased risk of gastric cancer, these changes, in particular intestinal metaplasia, are accepted as a marker of gastric cancer risk and may be interpreted as preneoplastic epithelial changes in gastric carcinogenesis. Progression to gastric cancer is higher in patients with more extensive forms of atrophic gastritis with intestinal metaplasia involving large areas of the stomach, including the gastric body and fundus. This pattern of gastritis has been described as pangastritis or multifocal atrophic gastritis.6,17 Extensive gastritis involving the gastric body and fundus results in hypochlorhydria, allowing for bacterial overgrowth and increased carcinogenic activity in the stomach through the conversion of nitrites to carcinogenic N-nitroso compounds. H. pylori-associated pangastritis is frequently seen in the relatives of gastric cancer patients, which may contribute to gastric cancer clustering in some families.18
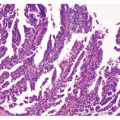
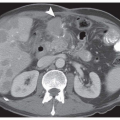
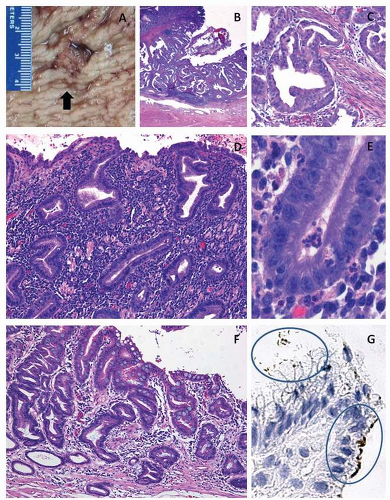 FIGURE 3-1 Classic findings in H. pylori-associated gastric carcinogenesis. A: Gross photograph of ulcerated adenocarcinoma in gastric antrum (arrow). B: Histology of ulcerated adenocarcinoma. C: Invasive moderately differentiated adenocarcinoma (intestinal type). D: Background mucosa reveals marked inflammation of the lamina propria (chronic gastritis). E: High-power view of gastric epithelium with associated neutrophils (active gastritis). F: Foci of intestinal metaplasia in the gastric mucosa affected by chronic gastritis. G: High-power view of H. pylori immunohistochemical analysis. H. pylori bacteria are present in the surface mucus (left-upper oval shape) and are attached to the apical aspect of gastric surface epithelial cells (right-lower oval shape), original magnification 400×. Hematoxylin and eosin stains original magnification 100× (B,C,F) and 200× (E). |
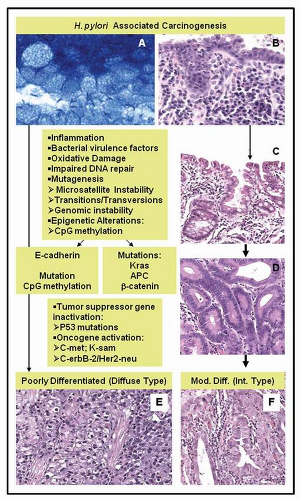 FIGURE 3-2 H. pylori-associated gastric carcinogenesis: alternative pathways and associated molecular patterns. A: H. pylori bacteria closely associate with the gastric epithelium (thiazine stain, 400×). B: Gastric mucosa with chronic active gastritis in response to H. pylori infection. C: Intestinal metaplasia stage of gastric carcinogenesis, progressing to dysplasia/adenoma (D) an invasive moderately differentiated (Mod. Diff.) adenocarcinoma of intestinal type (Int. Type) (F). E: Some cases of gastric cancer arising in association with H. pylori chronic gastritis do not progress through the stages of intestinal metaplasia and dysplasia, but rather abruptly present as poorly differentiated (diffuse type) adenocarcinomas. Hematoxylin and eosin stains original magnification 200× (B-F). |
Stomach cancers are classified according to the WHO classification on the basis of their grade of differentiation, which is categorized as well, moderately, and poorly differentiated
adenocarcinomas.19 The Lauren classification reported in 1965 has been widely used in studies of gastric adenocarcinomas, separating gastric cancer into the intestinal and diffuse types, on the basis of the morphologic features of the tumor. The cancers that arise in the inflammatory background of H. pylori-associated chronic gastritis are most commonly intestinal-type adenocarcinomas, which are predominantly well to moderately differentiated (Figs. 3-1 and 3-2), but diffuse-type tumors, which are poorly differentiated and may include a variable component with signet ring cell features, also occur in association with H. pylori9,20 (Fig. 3-2).
adenocarcinomas.19 The Lauren classification reported in 1965 has been widely used in studies of gastric adenocarcinomas, separating gastric cancer into the intestinal and diffuse types, on the basis of the morphologic features of the tumor. The cancers that arise in the inflammatory background of H. pylori-associated chronic gastritis are most commonly intestinal-type adenocarcinomas, which are predominantly well to moderately differentiated (Figs. 3-1 and 3-2), but diffuse-type tumors, which are poorly differentiated and may include a variable component with signet ring cell features, also occur in association with H. pylori9,20 (Fig. 3-2).
Inflammatory Cascades in H. pylori Chronic Gastritis
H. pylori infection of the stomach elicits a host inflammatory response that includes both humoral and cellular responses and involves both innate and acquired immune responses.16 Once H. pylori colonizes and infects the stomach, the disease persists as chronic gastritis, unless treatment is applied to eradicate the infection.15 Because of the long-term chronic inflammatory status of the gastric mucosa, H. pylori must be able to evade the immune system, and this evasion is thought to be the main contributor to gastric cancer development. The mechanisms that lead to H. pylori immune evasion and chronic gastritis are being unraveled. The immune response to H. pylori is induced when the bacterial products contact the gastric epithelial cells lining the stomach and the macrophages and dendritic cells in the lamina propria, which are reached after the epithelial cells and intercellular junctions are damaged by H. pylori virulence factors such as the vacuolating cytotoxin (VacA). This cellular damage permits the bacteria and bacterial products to move through the interrupted epithelial cell layer to the subepithelial lamina propria (reviewed in Ref. [16]). The steps involved in the host immune response to H. pylori can be described as follows: (a) H. pylori bacteria adhere to the apical aspect of epithelial cells that are primarily located on the mucosal surface and eventually move into the gastric foveolae, where the organisms may come into contact with gastric stem cells. The epithelial cells respond to H. pylori through cell-signaling events to produce cytokines, which are released into the lamina propria to activate macrophages, dendritic cells, and other inflammatory cells; (b) Some H. pylori bacteria are able to enter the lamina propria after the surface epithelial layer is damaged, allowing H. pylori to directly interact with innate and acquired immune response cells; (c) Macrophages, dendritic cells, and T-cell mediators released by the epithelial cells activate T lymphocytes with a predominant Th1 response, regulatory T lymphocytes (T-reg), B lymphocytes, which mature into mucosal plasma cells, and neutrophils, which actively phagocytize H. pylori. In addition, dendritic cells release IL23 and activate the production of IL17, which is associated with a Th17 response against H. pylori. Recent data indicate that H. pylori directs Treg-skewed dendritic cell-induced helper T-cell differentiation, in contrast to the Th17-skewed response seen with proinflammatory bacteria. The increased Treg induction in H. pylori-infected hosts forces an imbalance of the Th17/Treg axis, which may lead to ineffective bacterial eradication and the persistence of H. pylori as a chronic infection.21
Stem Cells and H. pylori-associated Gastric Carcinogenesis
Studies of H. pylori infection in mouse models have suggested a potential role for bone marrow-derived stem cells in chronic gastritis and H. pylori-associated neoplastic progression.22 The authors hypothesize that H. pylori-associated inflammation and glandular atrophy creates an abnormal microenvironment in the gastric mucosa that favors the engraftment of bone marrow-derived stem cells onto the inflamed gastric epithelium. The engrafted bone marrow-derived stem cells would not follow a normal differentiation pathway and would undergo uncontrolled replication, progressive loss of differentiation, and neoplastic behavior.22 However, the potential role of bone marrow-derived stem cells in human disease remains unclear.
H. pylori Strains and Virulence Factors in Gastric Carcinogenesis
H. pylori is characterized by a unique set of virulence factors that play significant roles in its pathogenesis and influence the course of H. pylori-associated diseases.23 The best known and most significant H. pylori virulence factors are VacA and the cytotoxin-associated antigen A (CagA)
proteins.24 Other bacterial factors that are important for H. pylori-related carcinogenesis include the BabA gene, which plays a role in bacterial adhesion to gastric epithelial cell blood group antigens. The combined presence of BabA, CagA and VacAs1 in H. pylori strains has been reported to be associated with duodenal ulcers and gastric adenocarcinomas in Western populations.
proteins.24 Other bacterial factors that are important for H. pylori-related carcinogenesis include the BabA gene, which plays a role in bacterial adhesion to gastric epithelial cell blood group antigens. The combined presence of BabA, CagA and VacAs1 in H. pylori strains has been reported to be associated with duodenal ulcers and gastric adenocarcinomas in Western populations.
VacA is an 88 kDa protein that is secreted by H. pylori and causes the vacuolization and apoptosis of epithelial cells. VacA has been shown to have immunomodulatory effects, affecting B-lymphocyte antigen presentation, regulation of the T cell-mediated cytokine response, inhibition of T-lymphocyte activation and proliferation, and inhibition of T lymphocyte-induced B-cell proliferation (reviewed in Ref. [5]). The immunomodulatory actions of the VacA toxin on T and B lymphocytes may contribute to the ability of H. pylori to establish persistent chronic gastritis.
The CagA protein is a 125 to 145 kDa protein encoded by the cagA gene, which is one of the genes that constitute the Cag pathogenicity island.24 This segment of the H. pylori genome encodes a type IV bacterial secretion system.24 Recent studies have elucidated the role of CagA in H. pylori pathogenesis. These studies include the characterization of the functional domains of the CagA protein, the demonstration that some structural forms are more virulent and that variants of this protein are produced by different H. pylori strains (reviewed in Ref. [24]). In addition, the mechanisms by which CagA affects epithelial cells and may contribute to gastric cancer development are becoming better understood.24 H. pylori strains carrying a cagA gene have been shown to have a stronger association with gastric cancer.23,25 H. pylori strains that produce the CagA protein are associated with an increased risk of gastric carcinoma, at least in part, because these strains cause greater inflammatory mucosal damage. CagA-positive strains have been shown to induce higher levels of interleukin-8 (IL-8) than CagA-negative strains, resulting in higher levels of inflammation in the gastric mucosa.26
Some structural variants of CagA have a stronger association with gastric cancer. The CagA type C strains are associated with more severe degrees of atrophic gastritis and gastric cancer. H. pylori strains with unique phosphorylation patterns of the CagA phosphorylation site in the amino acid motif EPIYA are more common in Eastern Asia than in Western countries, which may contribute to the increased incidence of gastric cancer in Eastern Asia.24




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
